以下内容,参考、并整理自授课教材:

文章目录
- [一,结构预测/结构建模(Structure Prediction/Computational protein structure modelling)](#一,结构预测/结构建模(Structure Prediction/Computational protein structure modelling))
-
- (一)4种结构预测方法
-
- [1,Comparative Modeling/Homology Modelling 比较建模/同源建模](#1,Comparative Modeling/Homology Modelling 比较建模/同源建模)
- [2,De Novo/Ab initio Modeling 从头预测](#2,De Novo/Ab initio Modeling 从头预测)
-
- 从头预测方法的定义
- 两种具体的从头预测方法
- [Rosetta vs TASSER](#Rosetta vs TASSER)
- 从头预测方法的优缺点
- [3,Threading 线程法/折叠识别](#3,Threading 线程法/折叠识别)
-
- [step1:准备工作 - 建立"试衣间"](#step1:准备工作 - 建立“试衣间”)
- [step2:关键动作 - "试穿"与"打分"](#step2:关键动作 - “试穿”与“打分”)
- [step3:做出决策 - "选择最合身的衣服"](#step3:做出决策 - “选择最合身的衣服”)
- [step4:成品加工 - "量身修改"](#step4:成品加工 - “量身修改”)
- 一个形象的比喻
- [1. 为什么需要Threading?------ 解决"同源建模"的局限](#1. 为什么需要Threading?—— 解决“同源建模”的局限)
- [2. 在概念上,Threading可以看作是同源建模的"逆过程"](#2. 在概念上,Threading可以看作是同源建模的“逆过程”)
- [4,Integrative(Hybrid)Modelling 整合/混合建模](#4,Integrative(Hybrid)Modelling 整合/混合建模)
- [(二)Scoring functions 打分函数](#(二)Scoring functions 打分函数)
-
-
- [1. 打分函数的定义和作用](#1. 打分函数的定义和作用)
- [2. 打分函数的四个主要组成部分](#2. 打分函数的四个主要组成部分)
- [3. 常见的打分函数类型](#3. 常见的打分函数类型)
- [4. 打分函数的应用](#4. 打分函数的应用)
-
- [(三)Model Assessment 模型评估](#(三)Model Assessment 模型评估)
- [二,结构分类/结构比较(Classification of Protein Structures/Structure Comparison)](#二,结构分类/结构比较(Classification of Protein Structures/Structure Comparison))
-
- [1,Structural alignment 结构比对](#1,Structural alignment 结构比对)
- 2,结构分类数据库
- [三, 分子动力学模拟Molecular Dynamics Simulations (MD)](#三, 分子动力学模拟Molecular Dynamics Simulations (MD))
- [四,分子对接(Docking of Ligands and Proteins/Molecular Docking)](#四,分子对接(Docking of Ligands and Proteins/Molecular Docking))
- [五,结构验证(Structure Validation)](#五,结构验证(Structure Validation))
- [六,虚拟筛选(Virtual screening)](#六,虚拟筛选(Virtual screening))
一,结构预测/结构建模(Structure Prediction/Computational protein structure modelling)
(一)4种结构预测方法
homology modelling, threading, ab initio modelling and integrative modelling
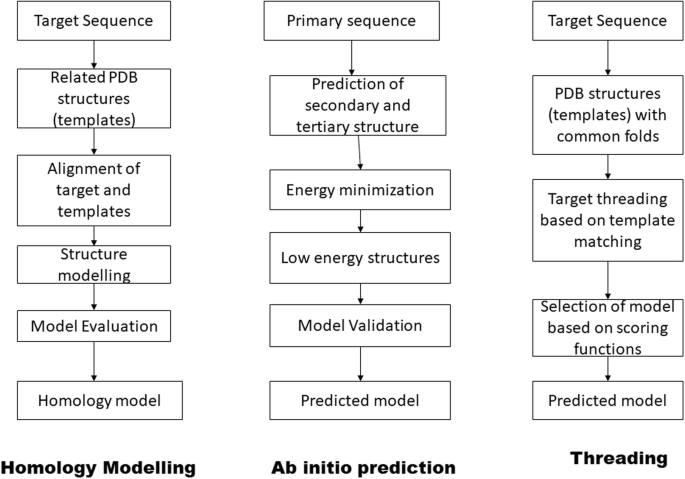
1,Comparative Modeling/Homology Modelling 比较建模/同源建模
evolutionary principle that protein sequences having common evolutionary descent are likely to have similar three-dimensional structures(具有共同进化血统的蛋白质序列可能具有相似的三维结构这一进化原则);
进化论(序列保守------》结构推断);
将目标蛋白的氨基酸序列与已知结构模板蛋白的氨基酸序列进行比较,通过比较靶序列与模板序列的相似性,计算算法可以预测靶蛋白的三维结构;
具有共同进化祖先的蛋白质表现出一种显著保守的三维构象,差异程度超过20%的蛋白质序列可能表现出不同的结构构象。
同源建模的定义和基本原理
- 定义:同源建模是一种基于进化原理的蛋白质结构预测方法。其核心思想是,具有共同进化起源的蛋白质序列,往往具有相似的三维结构。
- 基本原理:通过寻找与目标蛋白质(即需要预测结构的蛋白质)具有相似序列的已知结构蛋白质(称为模板蛋白质),利用这些模板蛋白质的结构信息来构建目标蛋白质的三维结构模型。
同源建模的具体步骤
- 模板选择:从蛋白质数据库(Protein Data Bank,PDB)中筛选出与目标蛋白质序列相似度较高、且结构已知的模板蛋白质。通常会挑选几个合适的模板结构,以便后续进行比较和选择。
- 序列比对:将模板蛋白质的序列与目标蛋白质的序列进行比对,确定它们之间的序列相似性。通过比对可以计算出模板与目标序列之间的序列一致性百分比(sequence identity)。一般来说,序列一致性越高,同源建模的准确性就越高。当序列一致性达到40%或更高时,模型的准确性较好,模型与模板结构之间的偏差通常小于2埃(2A RMSD)。但如果序列一致性低于30%,模型的准确性会大幅下降,容易出现较多的误差。
- 模型构建:根据序列比对的结果,以一个或多个可用的模板结构为基础,生成目标蛋白质的全原子模型。这个模型是基于模板结构的原子坐标信息构建的,会尽量保持模板结构的特征和空间排列。
- 模型优化:对生成的全原子模型进行优化处理,主要是通过能量最小化步骤来调整模型的原子位置,使其能量达到更低的状态,从而提高模型的稳定性和合理性。能量最小化可以减少模型中的不合理结构和能量冲突,使模型更加接近真实情况。
- 模型评估:使用一些专门的评分方法对优化后的模型进行整体几何准确性的评估。评估的目的是判断模型的三维结构是否合理、是否存在明显的结构错误等。如果序列一致性超过50%,通过同源建模可以得到一个非常好的模型,其主链原子的偏差小于1埃(1A RMSD)。而当序列一致性低于30%时,模型的准确性较低,可能会出现多种类型的结构错误。
同源建模的优缺点
- 优点:当存在与目标蛋白质序列相似度较高的已知结构模板时,同源建模可以快速、准确地构建出目标蛋白质的三维结构模型。这种方法相对较为成熟,且在许多情况下能够提供可靠的结构信息,为后续的生物学研究和药物设计等提供基础。
- 缺点:对模板蛋白质的依赖性较强。如果找不到合适的模板结构,或者模板与目标蛋白质的序列一致性过低,同源建模的准确性和可靠性就会大打折扣。此外,模型的优化和评估过程需要专业的软件和方法,对操作人员的专业知识和经验要求较高。
2,De Novo/Ab initio Modeling 从头预测
避免了预先存在的同源三维结构的要求,从序列直接获得三维结构;
从头预测方法的定义
- 不依赖模板结构:从头预测方法属于从新(de novo)方法的一种,它不依赖于已知的模板结构来预测蛋白质的结构。与同源建模不同,同源建模需要找到与目标蛋白有相似序列和已知结构的模板蛋白来进行建模,而从头预测方法则完全不依赖这样的模板。
- 基于能量函数或结构信息:从头预测方法依赖于能量函数或者从其他蛋白质结构推断出的信息来进行预测。它假设蛋白质的天然状态存在一个全局自由能最小值,这个假设是基于已知的蛋白质三级结构的构象空间以及氨基酸序列的低自由能状态来确定的。
两种具体的从头预测方法
- 1,Rosetta方法 :
- 利用短结构片段集合:该方法依赖于从蛋白质数据库(PDB)中获取的一系列短结构片段。这些短片段是蛋白质结构的局部信息,通过组合这些片段可以构建出完整的蛋白质结构。
- 蒙特卡洛搜索方法:采用蒙特卡洛搜索方法来根据评分函数组装这些结构片段。蒙特卡洛方法是一种基于随机抽样的计算方法,通过大量的随机尝试来寻找最优的结构组装方式,评分函数则用于评估组装后的结构的合理性。
- 应用范围:Rosetta方法被广泛应用于蛋白质结构的建模,尤其是对于那些结构变化较大的区域,如插入片段和环状结构等,这些区域在蛋白质的功能和稳定性中往往起着关键作用。
- 总的来说,就是:

- 2,TASSER方法 :
- 基于线程的方法:TASSER方法依赖于线程(threading)方法来寻找局部片段。线程方法是一种将目标序列与已知结构的蛋白质进行比对,以确定目标序列在已知结构上的最佳拟合位置的方法。
- 随机游走和精修过程:找到局部片段后,通过随机游走的方式将这些片段连接成全长模型,然后进行精修过程。随机游走是一种随机探索路径的方法,用于在可能的结构空间中寻找合理的连接方式,精修过程则是对初步构建的模型进行优化,以提高其准确性和合理性。
- 总的来说,就是:

Rosetta vs TASSER
1,Rosetta方法
- Rosetta方法:通过从PDB中提取短结构片段,使用蒙特卡洛搜索方法组装和优化这些片段,最终生成蛋白质结构模型。它更适合于小蛋白质的结构预测,但对计算资源需求较大。从PDB中提取短片段:
- 定义:从蛋白质数据库(PDB)中提取已知的短片段。这些短片段是已经存在于PDB中的蛋白质结构的一部分,是实际的、已知的结构片段。
- 目的:为后续的结构组装提供基础单元。这些短片段可以作为构建蛋白质三维结构的"积木",通过组合这些短片段来构建完整的蛋白质结构。
- 方法:通常会从PDB中筛选出具有特定长度和特征的短片段,这些片段可以是蛋白质的二级结构(如α-螺旋、β-折叠)或其他局部结构。提取这些片段后,会将它们存储在一个库中,供后续的结构预测方法使用。
- 例子:在Rosetta方法中,从PDB中提取长度为3到9个氨基酸的短片段,这些片段将被用于后续的结构组装过程。
2,TASSER方法
- TASSER方法:通过线程比对寻找局部片段,然后通过随机游走连接这些片段,并进行精修优化,最终生成蛋白质结构模型。它在处理大型蛋白质时可能更有优势,但也需要大量的计算资源。识别局部片段:
- 定义:识别蛋白质序列中的局部片段,这些片段可能与已知的短片段相似,但不一定是直接从PDB中提取的。识别的过程可能涉及序列比对、结构预测等方法,目的是找到蛋白质序列中可能具有特定结构特征的片段。
- 目的:为构建完整的蛋白质结构提供局部结构信息。这些局部片段可以是蛋白质序列中可能形成特定二级结构或局部三维结构的区域,通过识别这些片段,可以更好地预测蛋白质的整体结构。
- 方法:可以使用多种方法来识别局部片段,如基于序列的比对方法(如BLAST)、基于物理化学性质的预测方法(如PSIPRED)、或者基于机器学习的方法。这些方法可以帮助识别蛋白质序列中可能具有特定结构特征的片段。
- 例子:在TASSER方法中,通过线程(threading)方法识别蛋白质序列中的局部片段。这些局部片段可能与已知的短片段相似,但不一定是直接从PDB中提取的。识别这些局部片段后,会通过随机游走(random walk)和后续的优化过程来构建完整的蛋白质结构。

从头预测方法的优缺点
- 优点:从头预测方法能够对一些小蛋白质进行准确的结构预测,这在没有合适模板的情况下是非常有价值的。它为那些无法通过同源建模等依赖模板的方法来预测结构的蛋白质提供了一种可能的解决方案,有助于深入理解蛋白质的结构和功能。
- 缺点:从头预测方法存在一些固有的局限性。首先,它对计算资源的需求非常大,因为需要进行大量的计算来探索蛋白质的构象空间,寻找最优的结构。其次,对于大型蛋白质,从头预测方法所构建的模型质量往往较差,这可能是因为大型蛋白质的构象空间更加复杂,需要更多的计算资源和更精确的方法来探索和优化。
3,Threading 线程法/折叠识别
****其核心思想是:即使两个蛋白质的氨基酸序列相似性很低,它们也可能采用相同的三维折叠结构。
- 目标:为一个给定的目标氨基酸序列(未知结构)找到一个已知的、最合适的蛋白质三维结构(作为模板)。
- 方法:将目标序列的氨基酸"穿"或"贴"到候选模板结构的骨架上,通过一系列计算和评分函数来评估这种"穿戴"的合适程度,并找出最优匹配。(就好像是穿针引线,织衣服一样)
Threading 的核心思想是:"不管黑猫白猫,抓到老鼠就是好猫"。它不关心你的序列和模板序列在进化上是否相关(即是否同源),只关心你的序列"穿上"这个模板结构后"舒不舒服"。
step1:准备工作 - 建立"试衣间"
科学家们把所有已知的蛋白质三维结构(来自PDB数据库)收集起来,去除重复的,建立一个庞大的"服装库"或"试衣间"。这个库里的每一件"衣服"就是一个折叠模板。
step2:关键动作 - "试穿"与"打分"
这是Threading最核心的一步。对于你的目标序列(你的"身材数据"),计算机会把它依次"试穿"到模板库里的每一个结构上。
但这个"试穿"不是简单的套上去,而是一个复杂的计算过程,主要包括两个动作:
- 对齐 (Alignment) :
- 就像试衣服时要对准袖子、领口一样,计算机会把你的氨基酸序列(一串字母)和模板结构的空间位置进行对齐。
- 难点在于:由于序列相似度低,传统的序列比对方法(像BLAST)会失效。所以必须使用更高级的、考虑了结构信息的比对方法。比如,它会故意把疏水性的氨基酸对准模板结构内部疏水的区域。
- 打分 (Scoring) :
- 每对齐一次,系统就会计算一个分数 (或称伪能量),来评价这次"试穿"的效果有多好。你可以理解为"合身度评分"。
- 评分标准是什么? 主要看以下几点:
- 环境是否匹配? 疏水的氨基酸(怕水)是否被安排在了蛋白质分子的内部(避水)?亲水的氨基酸是否被安排在了表面(亲水)?如果是的,就加分;如果不是,就扣分。
- 二级结构是否匹配? 喜欢形成α螺旋的氨基酸(如谷氨酸、丙氨酸)是否被安排在了模板的螺旋段上?喜欢形成β折叠的氨基酸(如缬氨酸、异亮氨酸)是否被安排在了模板的折叠片上?如果是,就加分。
- 空间冲突检查: 两个在空间上被安排得很近的氨基酸侧链,会不会像两个撞在一起的衣架,产生冲突?如果有,就扣分。
- 空位罚分: 在序列比对中引入的空隙(Gap)就像衣服上的破洞,是要被惩罚扣分的。但惩罚的力度会灵活调整,比如在环区(loop)区域开个"破洞"就比在规整的螺旋区开"破洞"惩罚要轻。
step3:做出决策 - "选择最合身的衣服"
计算机会遍历完模板库中成千上万个模板,为每一个模板都计算出一个"合身度"分数。
- 最终目标 :找出那个分数最高(或能量最低,意味着结构最稳定)的模板。
- 这个得分最高的模板,就被认为是你的目标序列最可能采用的三维折叠方式。
step4:成品加工 - "量身修改"
选中了"最合身的成衣"(模板)后,并不是直接拿来穿。还需要进行加工:
- 构建模型:根据刚才最优的对齐方案,把目标序列的氨基酸搭建在模板的骨架上。
- 优化修饰:对模型进行能量优化,调整原子位置,消除不合理冲突。特别是对于那些序列差异很大的环区(Loop),需要进行重新建模。
- 质检:最后检查一下成品模型是否合理(比如键长、键角、侧链取向等)。
一个形象的比喻
| 步骤 | 服装店比喻 | Threading 的专业解释 |
|---|---|---|
| 1 | 进入大型服装商城 | 构建已知蛋白质结构的模板库 |
| 2 | 拿着自己的身材数据 | 输入目标氨基酸序列 |
| 3 | 拿起每一件衣服比划 | 将目标序列与每一个模板进行序列-结构比对 |
| 4 | 照镜子看是否合身 | 使用能量评分函数计算兼容性 |
| 5 | 选择最合身的那件 | 识别出评分最优的模板结构 |
| 6 | 对衣服进行微调 | 构建并优化三维模型 |
1. 为什么需要Threading?------ 解决"同源建模"的局限
要理解Threading的重要性,必须将其与另一种主要方法同源建模(Comparative Modelling) 对比。
- 同源建模 :
- 前提 :需要找到一个与目标序列高度同源(通常序列一致性 > 30%)的模板结构。
- 逻辑 :
序列高度相似 -> 结构必然相似。它基于进化相关性的推断。 - 局限:对于大量没有高同源模板的蛋白质(尤其是在人类基因组中),同源建模无法使用。
- Threading :
- 前提 :目标序列与模板序列相似性可以很低 (甚至低于20%),但只要它们折叠方式相同即可。
- 逻辑 :
结构相似 -> 功能可能相似。它跳过了直接的序列进化关系,直接比较序列与结构的兼容性。 - 优势:极大地扩展了结构预测的范围,能够为那些"孤儿"序列(没有明显同源物)找到可能的结构模板。
2. 在概念上,Threading可以看作是同源建模的"逆过程"
- 同源建模 :一个模板 vs 多个序列(寻找哪个序列最适合这个模板)。
- Threading :一个目标序列 vs 一个模板库(多个结构)(寻找哪个模板最适合这个序列)。
这个问题,就是如何从逻辑上理解同源建模与threading法的区别与联系,如何理解threading法可以看作是同源建模的逆过程,我曾经在结构生物信息学课上作为一道课后思考题,考过一些本科生。
初学者很容易混淆这里面的逻辑,因为同源建模和threading从实操步骤上看,都是从序列出发,所以无法理解这个"逆过程"到底应该如何理解?
核心区别在于搜索的起点和逻辑方向,而不是有没有从序列出发。

我们可以从查询对象和数据库的角度来理解:

4,Integrative(Hybrid)Modelling 整合/混合建模
这个就没什么好讲的了,可以看作是ensemble learning,集成学习;
- 定义:整合(混合)建模方法是一种计算方法,它将来自多种来源的结构信息(如X射线晶体学、核磁共振光谱学、冷冻电镜、交联结合质谱、序列分析等)结合起来,以获得大型蛋白质复合物的结构模型,该模型具有更高的精度、准确性和分辨率。
- 方法优势:这种方法利用每种方法的优势来相互补充,从而获得更好的结构模型。
- 迭代过程:整合建模通过迭代过程来进行,即先收集数据,然后根据数据提出一个初始模型,接着再次收集更多数据,以进一步完善和验证所提出的初始模型。
- 步骤 :
- 1:从各种实验方法和其他统计方法(如原子统计势和分子力学力场)中收集输入数据。
- 2:开发评分函数,以评估模型与输入数据的一致性。
- 3:生成多个模型,并使用各种优化算法(如蒙特卡洛算法、遗传算法和梯度下降算法)来提高它们的评分。经过优化后,得分较高的模型代表了系统的构象。
- 4:将所有得分较高的模型进行聚类和分析,以获得整个模型集合的总体精度和准确性。此外,它还可以为下一次迭代中可能进行的实验提供线索。
(二)Scoring functions 打分函数
其实打分函数的这个概念学计算生物学的应该不陌生了,在序列比对这个最原始最入门的话题上,我们也能够看到打分矩阵、打分函数的思想理念在。
结构生物学这一块,在译名上其实有很多同类说法,打分函数、评分函数、能量函数、势能函数等等,暂时不作区分。
1. 打分函数的定义和作用
- 定义:打分函数是用于蛋白质结构预测的多维矩阵。它们基于势能函数来预测评估蛋白质的三维结构。
- 作用 :
- 评估实验确定的和计算机预测的蛋白质结构。
- 考虑原子间相互作用和溶剂化效应,因此也被称为有效能量函数。
2. 打分函数的四个主要组成部分
- 实体定义(Body Definition) :
- 可以由单个原子、一组原子的质心,或具有共同物理化学性质的不同原子或质心组成。
- 例如,定义20种标准氨基酸的α碳原子。
- 几何描述符(Geometrical Descriptor) :
- 描述不同实体之间的相互作用,使用一些度量,如:
- 角度
- 成对距离
- 二面角或扭转角
- 径向或角密度
- 描述不同实体之间的相互作用,使用一些度量,如:
- 参考系统(Reference System) :
- 是系统所有可能状态的加权平均状态。
- 用作参考设置,以计算特定状态的特定分数。
- 限制条件(Set of Restraints) :
- 定义分数的上限和下限。
- 或将分数划分为需要特定处理的不同类别。
3. 常见的打分函数类型
- 接触评分函数(Contact Scoring Functions) :
- 最简单的评分函数形式。
- 矩阵尺寸小,包含的实验数据较少。
- 例如,接触势能是一个20行20列的平方二维矩阵,用于定义20种标准氨基酸的α碳原子。
- 通常对称地为标准氨基酸的β碳原子或侧链质心推导。
- 距离依赖评分函数(Distance-Dependent Scoring Functions) :
- 在蛋白质结构预测中应用最为广泛。
- 由一个四维矩阵表示:
- 前两个维度:定义的实体总数。
- 第三个维度:区分局部和非局部相互作用。
- 第四个维度:描述距离范围或区间,以便表示相互作用。
- 可及表面评分函数(Accessible Surface Scoring Functions) :
- 捕捉定义实体与溶剂相互作用的倾向性。
- 描述在残基或原子水平上的溶剂可及性。
- 组合评分函数(Combined Scoring Functions) :
- 结合了可及表面和接触加距离依赖方法的信息。
- 接触和距离依赖方法处理蛋白质分子内的相互作用------》intra。
- 可及表面方法处理溶剂与蛋白质之间的相互作用------》inter(蛋白质和溶剂,比如说溶剂化效应之类的)。
- 需要一个归一化步骤将这两个独立的评分函数结合起来。
4. 打分函数的应用
- 模型选择 :
- 从不同的替代模型中选择出最准确的模型。
- 折叠评估 :
- 使用评分函数对预测模型进行折叠评估,以评估正确的折叠。
- 几何准确性评估 :
- 评估整体模型以及各个区域的几何准确性。
- 稳定性预测 :
- 在突变筛选的情况下,使用评分函数预测蛋白质结构的稳定性。
(三)Model Assessment 模型评估
模型评估的主要目标是评估预测模型的总体准确性。此外,它还从一组可供选择的模型中选择最精确的模型,并评估模型不同区域的精度。
模型评估的目的
- 总体准确度评估:模型评估的主要目标是评估预测模型的整体准确度(比如说Alphafold系列的pTM、ipTM等指标)。
- 模型选择:从多个备选模型中选择最准确的模型(同理,AF预测多个结果中model 0 cif为最高置信度)。
- 区域准确度评估:评估模型不同区域的准确度(同理,AF的pLDDT、PAE等指标)。
常用的model评价方法有4种:physics-based energies, knowledge-based potentials, combined scoring functions and clustering approaches。
四种常见模型评估方法
- 基于物理的能量方法(Physics-based energies)
- 化学力场(Chemical force fields):化学力场是系统中粒子的基于物理的能量函数,它来源于量子力学计算和实验结果。
- 能量函数的组成:力场能量函数由相互作用原子之间的化学键能量(如距离、角度和二面角)以及非键原子之间的相互作用(如静电和范德华力)组成。
- 应用:这些基于物理的方法在从一组预测模型中选择接近天然结构的模型方面很有用。
- 基于知识的势能方法(Knowledge-based potentials)
- 基于经验观察:这种方法基于对已知蛋白质结构的经验观察。
- 实现形式:以统计势能或平均力势能的形式实现。
- 统计势能类型:包括接触势能、距离势能、溶剂可及性势能以及成对相互作用势能等,这些势能用于评估模型的结构特征。
- 组合打分函数(Combined scoring functions)
还是ensemble learning那一套,集成学习。
- **优化加权组合**:组合打分函数是物理方法和基于知识的方法的各种分数的优化加权组合。
- **局限性**:然而,所有三种打分方法(基于物理的能量方法、基于知识的势能方法和组合打分函数)通常都难以检测到接近天然结构的模型。- 聚类方法(Clustering approaches)
- 模型生成与比较:从同一序列生成多个模型,并在聚类方法中对这些模型进行结构比较,以选择最佳模型。
- 打分函数质量:用于生成构象集合的打分函数的质量决定了模型的准确度。
- 统计概率转换:将可能构象集合中有用的信息转换为统计概率,以便识别蛋白质的近天然结构。
- 局限性:然而,聚类方法不能单独评估模型的质量,需要借助其他打分函数。
二,结构分类/结构比较(Classification of Protein Structures/Structure Comparison)
这里其实和前面的序列比较是呼应的,而序列比较,也就是序列比对,其实是计算生物学中最基础、入门的部分。
相应的评估方法、比对算法,优化方法,以及打分函数、打分矩阵,都是基础中的基础,这部分不是结构生物学的重点,故略。
1,Structural alignment 结构比对
通过评估三维结构的形状和构象来促进三维结构的比较,相同位置的原子被旋转和平移,这样一种结构就可以叠加在另一种结构上
(1)Root mean square deviation (RMSD) 均方根偏差
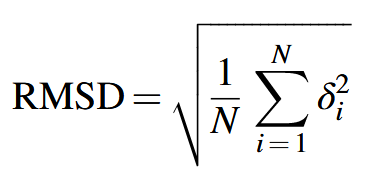
用均方根偏差(RMSD)来测量两个重叠蛋白质中等效原子之间的距离。
(2)蛋白质结构分类的基础:
- 蛋白质结构分类主要是基于对单个结构域的识别以及它们的进化关系。这是因为蛋白质的结构和功能往往与其中的结构域密切相关,而进化关系则反映了蛋白质在进化过程中结构和功能的保守性或变化。
- 评估两个蛋白质之间的结构相似性通常是通过将一个蛋白质结构中的Cα原子(Cα原子是蛋白质主链上的碳原子,位于氨基酸残基的中心位置,常用于代表氨基酸残基的位置)与另一个蛋白质结构中的Cα原子进行叠加,然后测量两个叠加蛋白质中对应原子之间的距离。这种距离是用均方根偏差(RMSD)来衡量的。RMSD是一种统计学方法,用于量化两个数据集之间的差异。在蛋白质结构分析中,它表示两个蛋白质结构在空间上的平均偏差。如果RMSD值小于3.5 Å(Å是埃,1 Å = 0.1纳米,是衡量原子间距离的常用单位),则表明这两个蛋白质在结构上非常相似,进而可能在结构上存在同源性(即它们可能在进化上具有共同的祖先)。(3)Graph-Based Structural Signatures
Structural signatures/fingerprints结构特征、指纹,大分子模式的表征
2,结构分类数据库
目前只有两个人工管理的蛋白质结构分类数据库,即CATH数据库和涵盖PDB中全部蛋白质结构的SCOP数据库。
https://www.ebi.ac.uk/pdbe/scop/
- CATH数据库
- CATH数据库是一个经过人工精心整理的蛋白质结构分类数据库,它涵盖了PDB中存储的所有蛋白质结构。CATH代表了四个层级的分类体系,分别是:
- Class(类别):这是最顶层的分类,主要根据蛋白质的二级结构含量来划分。例如,主要是α-螺旋结构、主要是β-折叠结构和混合的α-β结构。二级结构是指蛋白质主链原子局部的空间排列,α-螺旋和β-折叠是两种常见的二级结构形式。
- Architecture(架构):进一步将蛋白质分为40个组,依据是二级结构的总体布局,而不考虑它们在三维空间中的连接方式。这就像从宏观角度观察蛋白质的形状和结构布局。
- Topological motif(拓扑基序):将蛋白质分类为1000多个拓扑基序或折叠群。这一层级的分类不仅考虑了二级结构的排列,还考虑了它们之间的连接方式。也就是说,它更细致地分析了蛋白质的三维空间结构。
- Homologous superfamily(同源超家族):最后,将结构域聚类为2000多个同源超家族。这个分类是基于它们的进化关系,即这些结构域在进化过程中具有共同的祖先。这些结构域不仅在结构上具有显著的相似性,而且在序列(即氨基酸序列)和/或功能上也有相似之处。
- CATH数据库在分类结构域时,采用自动化算法和人工整理相结合的方法。这种方法综合考虑了序列、结构和功能的相似性,以确保分类的准确性和可靠性。自动化算法可以快速处理大量的数据,而人工整理则可以纠正算法可能产生的错误,保证分类的质量。
- CATH数据库是一个经过人工精心整理的蛋白质结构分类数据库,它涵盖了PDB中存储的所有蛋白质结构。CATH代表了四个层级的分类体系,分别是:
- SCOP数据库
- SCOP数据库也像CATH数据库一样,将蛋白质结构划分为一个或多个结构域,但整个分类过程是纯手工完成的。这意味着它依赖于专家的经验和判断来进行分类。
- SCOP数据库遵循多级分类体系,最高层级是类别,同样是根据蛋白质的二级结构含量来安排。它包含五个主要类别:
- all alpha(全α):蛋白质主要由α-螺旋组成。
- all beta(全β):蛋白质主要由β-折叠组成。
- alpha/beta(α/β):α-螺旋和β-折叠相互交错。
- alpha+beta(α+β):α-螺旋和β-折叠是分开的,没有相互交错。
- multidomain(多结构域):包含两个或多个来自不同类别的结构域。
- 与CATH数据库不同的是,SCOP数据库中没有架构层级。不过,它也包含了折叠群和超家族组,这与CATH数据库类似。折叠群和超家族组都是基于蛋白质结构和进化关系的分类。
三, 分子动力学模拟Molecular Dynamics Simulations (MD)
研究分子及其组成原子在特定时间框架内的动态行为来模拟和分析分子系统,阐明系统行为和轨迹确定;
依赖于牛顿运动方程的应用,加上分子力学的方法,来估计粒子间的力,通常被称为力场(force field)。
分子动力学模拟,是研究复杂系统中分子间相互作用,也就是是基于网络、研究复杂网络的model;
model本身是基于力场的,大多数也是使用牛顿物理学定律的简单近似来模拟原子运动。
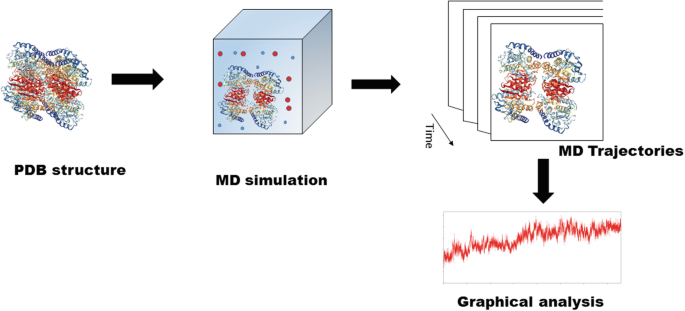
1,分子动力学模拟的原理
- 与分子间相互作用的联系:分子动力学模拟与复杂系统中分子间相互作用的概念密切相关。它通过模拟分子的运动来研究分子间的各种相互作用过程,例如蛋白质折叠、蛋白质-配体相互作用和蛋白质动力学等。
- 基于力场的模型:这些模拟模型是基于力场的。力场是一种描述分子中原子间相互作用的数学模型,它通过定义各种能量项来描述原子间的相互作用力,从而为模拟提供基础。
- 牛顿力学的近似应用:分子动力学利用对牛顿物理定律的简单近似来模拟原子的运动。这意味着它将原子视为质点,并根据牛顿运动定律来计算原子在力的作用下的运动轨迹。------》牛顿力学那一套就不说了,经典力学最基础的,应该是中学生都懂的那一套
2,分子动力学模拟的过程
- 初始模型的获取:模拟的初始模型通常来源于实验结构,如核磁共振(NMR)和X射线晶体学得到的结构,或者通过计算同源建模得到的结构。
- 力的估算 :系统中每个原子所受的力是通过一个势能函数方程来估算的。这个方程描述了原子间相互作用产生的力,包括键合原子(如共价键连接的原子)和非键合原子(如通过范德华力或静电力相互作用的原子)之间的相互作用。
- 能量函数的组成:总能量函数由多个部分组成,包括键长势能项(Eb)、键角势能项(Eθ)、二面角(扭转)势能项(Eφ)、非键合范德华相互作用项(EvdW)和非键合静电项(Eelc)。
- 基本上是分成两部分,成键原子之间的相互作用,以及非成键原子之间的相互作用,基本上所有的教材描述的都是(interactions between bonded and non-bonded atoms);前者一般依据化学描述符,一般比如说描述的是键长、键角、二面角,后者一般是范德华力、静电相互作用。
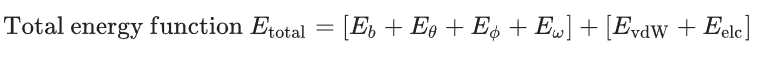
- **可选的约束项**:除了这些能量函数外,有时还会施加一些约束,以使分子处于期望的构象,作为总势能函数的可选项。3,力场在分子动力学模拟中的作用和地位
力场(force filed),在分子动力学模拟中是核心概念,简单来理解力场,其实可以认为是一系列数学规则,就是用来描述分子中原子之间应该如何相互作用的规则;
不同的model,其实拟合的是不同的一组规则,所以在某种程度上,我们其实可以将不同的力场看成是不同的model,也就是将力场视为描述分子中原子间相互作用的model,只不过这个model更加抽象、更加复杂。
我们拿到的其实是元model(meta-model),具体施加在这个复杂网络体系中每一个原子上面的力,都需要从meta-model再进一步解码。

从机器学习的角度来看,分子动力学中的力场确实可以被视为一种"模型",而模型的核心就是参数。这种类比可以帮助我们更好地理解力场的作用和优化过程。
(1)力场作为模型
在分子动力学中,力场(force field)定义了原子间相互作用的数学形式和参数,这些参数决定了原子如何在力的作用下运动。从机器学习的角度来看,力场可以被视为一个模型,它通过一系列参数来描述分子系统的动力学行为。
- 参数化模型:力场中的参数(如键长、键角、范德华半径、部分电荷等)类似于机器学习模型中的权重和偏置。这些参数决定了模型的行为,即原子间的相互作用力。
- 模型的优化:在机器学习中,模型的优化过程是通过调整参数来最小化损失函数。在分子动力学中,力场的优化过程是通过调整参数来拟合实验数据或量子力学计算结果。
(2)从贝叶斯学派与频率学派的角度来理解力场model
贝叶斯学派
贝叶斯学派认为模型参数是随机变量,具有先验分布。通过观察数据,可以更新这些参数的分布,得到后验分布。这种方法强调了参数的不确定性,并通过贝叶斯定理来更新参数。
- 先验分布:在分子动力学中,先验分布可以类比为基于物理直觉或先前实验数据的初始参数设置。例如,某些化学键的键长和键角可以根据已知的化学知识进行初步设定。
- 后验分布:通过模拟结果与实验数据的对比,可以更新这些参数的分布。例如,如果模拟结果与实验数据不符,可以通过调整参数来改善拟合效果。
- 贝叶斯优化:贝叶斯方法可以用于力场参数的优化。通过构建一个概率模型来描述参数与模拟结果之间的关系,可以更有效地搜索参数空间,找到最优的参数组合。
频率学派
频率学派认为模型参数是固定的,但未知的。通过观察数据,可以估计这些参数的值。这种方法强调了参数的点估计和置信区间。
- 点估计:在分子动力学中,点估计可以类比为通过最小化误差函数来确定最佳参数值。例如,通过最小化模拟结果与实验数据之间的均方误差来调整力场参数。
- 置信区间:频率学派还关注参数估计的置信区间,这可以帮助评估参数估计的可靠性。在分子动力学中,可以通过交叉验证等方法来评估参数的稳定性。
- 频率优化:频率方法通常依赖于梯度下降等优化算法,通过迭代调整参数来最小化误差函数。这种方法在实际应用中非常常见,例如在AMBER和GROMACS等软件中,通常使用这种方法来优化力场参数。
(3)力场的作用
- 描述原子间的相互作用 :
- 力场通过定义一系列能量项来描述分子中原子之间的相互作用。这些能量项包括键长、键角、二面角、范德华力和静电力等。这些能量项共同决定了原子之间的力,从而影响原子的运动。
- 例如,键长势能项描述了原子之间通过共价键连接时的能量变化,键角势能项描述了三个原子之间的角度变化,二面角势能项描述了四个原子之间的扭转角度变化。
- 非键合相互作用,如范德华力和静电力,则通过Lennard-Jones势和库仑定律来描述。
- 提供计算基础 :
- 力场为分子动力学模拟提供了计算基础。通过力场,可以计算出每个原子所受的力,进而根据牛顿运动定律更新原子的位置和速度。
- 力场的参数化过程确保了这些能量项能够合理地反映真实分子的物理和化学性质。
- 平衡计算精度和效率 :
- 力场通过简化的经典力学模型来描述复杂的量子力学现象,从而在保持一定精度的同时,显著提高了计算效率。这使得分子动力学模拟能够在合理的时间内处理大规模的分子系统。
(4)力场的地位
- 核心组件:力场是分子动力学模拟的核心组件。没有力场,就无法计算原子之间的相互作用力,也就无法进行分子动力学模拟。
- 桥梁作用:力场在经典力学模型和量子力学模型之间起到了桥梁的作用。它通过参数化过程,将量子力学计算得到的精确数据转化为经典力学模型中可以使用的参数。
- 可扩展性:力场的参数化方法使得它可以根据不同的分子系统和研究目的进行调整和优化,具有很强的可扩展性。
(5)为什么使用牛顿力学而不是量子力学
这个问题其实是很自然的想法,毕竟经典力学的局限性,是中学生学物理时就已经耳熟能详的了;
而量子力学的入门,也是中学生阶段就已经接触到了的;
所以这个问题,其实是一个中学生都能够质疑的问题。
那么为什么我们常见的力场一般都是使用经典力学,而不是使用更精确的量子力学呢?(从物理学角度上,确实量子力学更加形而上学,更接近于真实物理世界)。
The actual behaviour of real molecules in motion can only be modelled after parameterizing the energy terms to fit quantum mechanical calculations(真正分子在运动中的实际行为只能在参数化能量项后才能模拟,以符合量子力学计算)。
量子力学的局限性
- 计算复杂度 :
- 量子力学计算,如从头算(ab initio)方法或密度泛函理论(DFT),虽然能够提供非常精确的结果,但计算复杂度极高。对于大规模的分子系统(如蛋白质、核酸等生物大分子),这些方法的计算成本几乎是不可承受的。
- 例如,一个包含数千个原子的蛋白质分子,使用量子力学方法进行全系统的计算可能需要数年甚至数十年的计算时间,即使在超级计算机上也是如此。
这个其实也在情理之中,数值分析那一套,其实要回归到计算机数学上(这个不在我范畴之内,暂时略)。
但直观上讲,解一个经典力学的常微分方程,其实要比解一个薛定谔方程要容易得多;
真要解一个复杂点的偏微分方程,A4纸要写3~4页满的那都是家常便饭。
- 适用范围 :
- 量子力学方法通常适用于小分子系统或特定的化学反应过程。对于大规模的生物分子系统,量子力学方法的适用范围有限,且难以处理复杂的溶剂环境和动态过程。
牛顿力学的优势
- 计算效率 :
- 牛顿力学通过简化的经典力学模型来描述原子的运动,计算效率高。这使得分子动力学模拟能够在合理的时间内处理大规模的分子系统。
- 例如,使用分子动力学模拟,可以在数小时到数天内模拟一个包含数千个原子的蛋白质分子的动态过程,这在量子力学方法中是难以实现的。
- 适用范围广 :
- 牛顿力学模型适用于大规模的生物分子系统,能够处理复杂的溶剂环境和动态过程。通过力场的参数化,可以将量子力学计算得到的精确数据转化为经典力学模型中可以使用的参数,从而在保持一定精度的同时,显著提高了计算效率。
- 可扩展性 :
- 牛顿力学模型具有很强的可扩展性。通过调整力场的参数,可以适应不同的分子系统和研究目的。
总的来说,这是一个计算精度和效率的平衡,而trade-off在理想化的计算和真实物理世界的复杂性之间是非常常见的。
4,分子动力学模拟的建模方式
- 化学键和原子角度的建模:化学键和原子角度是基于简单的虚拟弹簧来建模的,分别对应键的伸缩和角度的弯曲。
- 二面角的建模:二面角(围绕键的旋转)是通过正弦函数来建模的。
- 非键合力的建模:非键合力,如范德华相互作用和静电相互作用,分别通过Lennard-Jones势和库仑定律来建模。
- 参数化的重要性:要模拟真实分子的运动行为,需要对能量项进行参数化,以使其符合量子力学计算的结果。参数化过程包括确定代表化学键和原子角度的弹簧的理想刚度和长度、估算用于计算静电相互作用能量的最佳部分原子电荷、确定合适的范德华原子半径等。所有这些参数统称为力场,因为它们分别对控制分子动力学的原子力做出了贡献。
5,常用的分子动力学模拟软件
- GROMOS、AMBER、CHARMM和GROMACS:这些都是常用的力场,它们在参数化方式上有所不同。这些软件提供了进行分子动力学模拟所需的工具和算法。
总结如下:
另外补充一个我常用的,也是高性能、定制化的,用于分子模拟的工具,OpenMM(仅代表个人观点 ):
https://openmm.org/
https://github.com/openmm/openmm
6,分子动力学模拟的执行
- 原子位置的更新:在估算出系统原子所受的力之后,根据牛顿运动定律来移动原子的位置。本质上就是牛顿第二定律,然后根据运动学方程更新原子的速度和位置。
- 时间步进的重复:通过将模拟时间反复向前推进一小部分秒(通常为飞秒级别),重复数百万次,来模拟分子的运动过程。
1️⃣推进模拟时间
在分子动力学模拟中,时间是离散的,通过一个小的时间步长 ( Delta t ) 来逐步推进。通常,时间步长非常小,例如在飞秒(10^-15秒)级别。每次更新原子位置后,时间向前推进 ( Delta t )。
这个过程会重复数百万次,以模拟足够长的时间,从而观察分子系统的动态行为。例如,如果时间步长是1飞秒,模拟1纳秒(10^-9秒)的时间就需要进行1000次迭代。
2️⃣重复迭代
整个过程是一个循环:
- 计算当前时间步中每个原子所受的力。
- 根据牛顿运动定律更新原子的速度和位置。
- 将时间向前推进一个时间步长 ( Delta t )。
- 重复上述步骤,直到达到预定的模拟时间。
- 计算资源的需求:由于需要对大量的原子进行力的计算和位置更新,并且这个过程需要重复数百万次,因此分子动力学模拟通常需要大量的计算资源。这就是为什么这些模拟通常在高性能计算机集群或超级计算机上进行。
7,分子动力学模拟结果的分析
- 标准图表的应用 :MD模拟的输出结果通常使用标准图表进行分析,如均方根偏差(RMSD)和均方根涨落/波动(RMSF)。
- RMSD:RMSD是衡量原子在模拟过程中随时间的位置偏差的指标,它反映了分子结构在模拟过程中的稳定性或变化程度。


评估的是一定时间内,整个model的原子坐标的变化指标,当然归一化到单个原子的坐标变化。
比如说下面的这个时间序列图,横轴表示模拟时间,纵轴表示RMSD值,随着时间变换,突变肽位置收敛到参考肽位置。
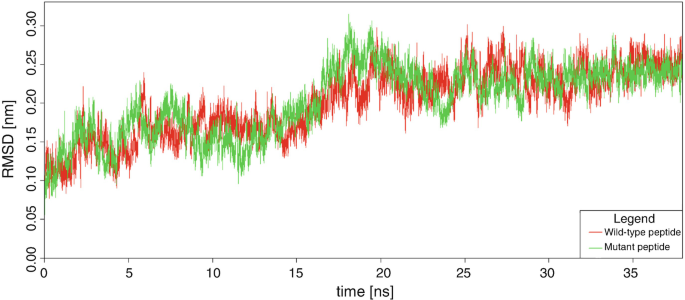
- **RMSF**:RMSF表示某个原子在一定时间内的位置变化程度,用于评估分子中不同部分的灵活性或刚性。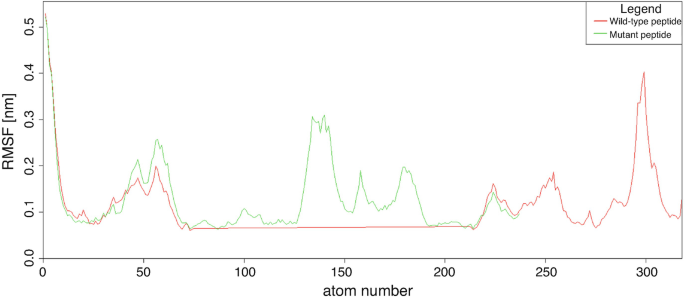
评估的是一个原子,在指定时间步内(所有时间步)的坐标的变化指标,归一化到单位时间内该原子坐标的变化值。
四,分子对接(Docking of Ligands and Proteins/Molecular Docking)
一种寻找配体与特定蛋白质结合的最佳适合方向的优化问题,
预测配体分子与受体或靶分子结合后的精确空间排列和坐标,
预测配体与受体上不同区域结合时潜在构象的一种计算方法,也称为结合模式;
应用利用分子对接算法,利用力场原理确定客观评分,从而促进最佳分子构象的排序,这种评分机制被设计用于优先考虑在研究的两个生物分子之间表现出增强的分子间相互作用的姿势;
预测微小化合物与蛋白质之间复杂的相互作用,破译大分子中复杂连接和结合模式。


1,对接的重要性
- 药物作用机制的关键:在水溶液中,小分子配体或蛋白质与它们的蛋白质受体之间的相互作用,对于理解药物活性化合物的作用机制至关重要。
- 奠定药物设计基础:配体与受体之间的这种相互作用被称为对接,它为基于结构的计算机辅助药物设计奠定了基础。
2,对接的原理
- 多种相互作用的稳定作用:配体-受体复合物不仅通过分子间的范德华力、氢键和静电相互作用来稳定,还通过分子表面非极性部分的脱溶作用来稳定。在结合之前,配体和受体都与溶剂相互作用。非极性表面部分的脱溶会释放水分子,导致熵的增加,从而进一步加强复合物的形成。
- 结合自由能的贡献因素:蛋白质-配体的结合亲和力以自由能增益来衡量。自由能增益的最大贡献来自于受体疏水区域中水分子的置换。配体与受体蛋白之间的氢键是配体结合后另一个重要的自由能来源。这种自由能增益是通过配体置换与受体蛋白结合的水分子来实现的。
- 结合自由能公式:描述配体与受体结合过程中自由能的变化
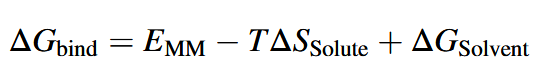

3,对接的方法
- 对接程序的两个重要组成部分 :
- 对接算法:用于搜索构型和构象的自由度,倾向于通过广泛搜索势能面来寻找全局能量最小值。在刚性对接中,只允许配体与受体活性位点之间有平移和旋转自由度。相比之下,柔性配体对接涉及配体的扭转自由度,允许配体二面角发生变化。注意一下柔性对接,与刚性对接。
- 打分函数:通常不仅评估配体与其受体之间的立体互补性,还评估它们的化学互补性。
- 以Autodock为例的对接过程:Autodock是一种流行的对接程序,它实现了具有数万步的模拟退火蒙特卡洛算法。首先,将配体随机放置在受体的结合位点上,并允许其向全局能量最小值移动。每次移动后,结构都会被最小化,随后测量新结构的能量。模拟可能包括几个周期,每个周期的温度逐渐降低。前一个周期的最低能量成为下一个周期的起始点。
- 结合自由能公式与分子对接中的打分函数的关系:共同构成了分子对接过程中评估配体与受体结合亲和力的核心框架:
从公式的组成形式上看,初学者很容易混淆结合自由能公式和打分函数。
总的来说,结合自由能公式提供了配体与受体结合的理论框架,而打分函数则是这一理论在分子对接中的具体应用。打分函数通过评估配体与受体之间的相互作用能量,预测结合亲和力,从而指导药物设计和优化,两者相辅相成。
打分函数用于评估配体与受体结合的亲和力,其目的是预测配体与受体结合的稳定性和结合自由能。通常包含以下几个部分:

具体来说,结合自由能公式与打分函数的联系:
- 能量项的对应关系 :
- 范德华力和静电相互作用:打分函数中的范德华力和静电相互作用项直接对应于结合自由能公式中的 ( E_MM ) 部分。
- 氢键:氢键项在打分函数中通常被视为一种特殊的范德华力和静电相互作用的组合,也与 ( E_MM ) 相关。
- 脱溶效应:打分函数中的脱溶效应项对应于结合自由能公式中的 ( Delta G_solvent ) 部分。
- 熵的考虑 :
- 打分函数中的熵:虽然打分函数通常不直接计算熵变 ( Delta S_solute ),但通过优化配体的构象和结合位点,间接考虑了熵的影响。例如,允许配体在结合过程中有一定的构象自由度,可以部分补偿熵的损失。
- 全局能量最小值 :
- 对接算法的目标:对接算法的目标是找到配体与受体结合的全局能量最小值,这与结合自由能公式的目标一致。通过最小化结合自由能,可以预测最稳定的配体-受体复合物结构。
在分子对接中的作用
- 预测结合亲和力 :
- 打分函数的作用:打分函数通过评估配体与受体之间的相互作用能量,预测配体与受体结合的亲和力。结合自由能公式提供了理论基础,打分函数则是这一理论在实际计算中的具体实现。
- 优化配体结合:通过打分函数,可以评估不同配体构象和结合位点的能量,从而优化配体的结合方式,提高结合亲和力。
- 指导药物设计 :
- 虚拟筛选:在药物设计中,打分函数用于虚拟筛选大量的配体,预测哪些配体可能与受体有较强的结合亲和力,从而缩小实验验证的范围。
- 结构优化:通过打分函数评估配体与受体的结合情况,指导配体的结构优化,提高药物的活性和选择性。
4,对接程序的评估
- 准确性评估方式:通常通过将预测的配体-受体复合物结构与通过X射线晶体学获得的实验结构进行比较来评估对接程序的准确性。在这样的比较中,每个配体都对接到其自身的同源受体构象(自对接)。有时,还会通过将配体对接到非同源受体构象来测试程序的准确性,例如受体的无配体结构或与不同配体共结晶的结构。这种测试对接程序准确性的方法被称为交叉对接。
- 评分函数的构成:评分函数一般由范德华相互作用、氢键和静电相互作用的三个力场项以及代表脱溶效应的项组成。
五,结构验证(Structure Validation)
验证策略被用于确定计算模型的准确性和可靠性
六,虚拟筛选(Virtual screening)
用于加快大量化合物文库的筛选过程,从而促进潜在候选药物的发现;
利用对接算法的力量,根据小分子与特定靶受体的亲和力来优先排序小分子