在当前的计算生物学与AI制药领域,精准调控基因表达、开发新型核酸药物以及优化基因编辑工具(如CRISPR系统),一直是学术界与产业界攻坚的核心。"蛋白质-核酸"相互作用是生命活动的基础,而如何从头设计出能够与目标特定核酸序列(DNA/RNA)高亲和力结合的多肽分子,则是这一领域的关键技术瓶颈。
随着AlphaFold3在复杂生物大分子互作预测上的突破,以及基于全原子扩散架构的生成模型(如Boltzgen)的问世,我们已经拥有了一套高效的计算生物学解决方案。
近期,国内前沿的生物技术服务商**科晶生物** 依托这些最新算法,跑通了一套成熟的**"从头设计核酸结合多肽"**计算机辅助管线。通过"靶点构象分析-生成式设计-分子对接初筛-精确建模验证"的漏斗式筛选闭环,大幅提升了核酸结合多肽设计的成功率与可靠性。
今天,我们就来拆解这一极具工程美感的前沿技术流程,看看一条高活性的"多肽结合物(Binder)"是如何在硅基数字世界中被"打磨"出来的。
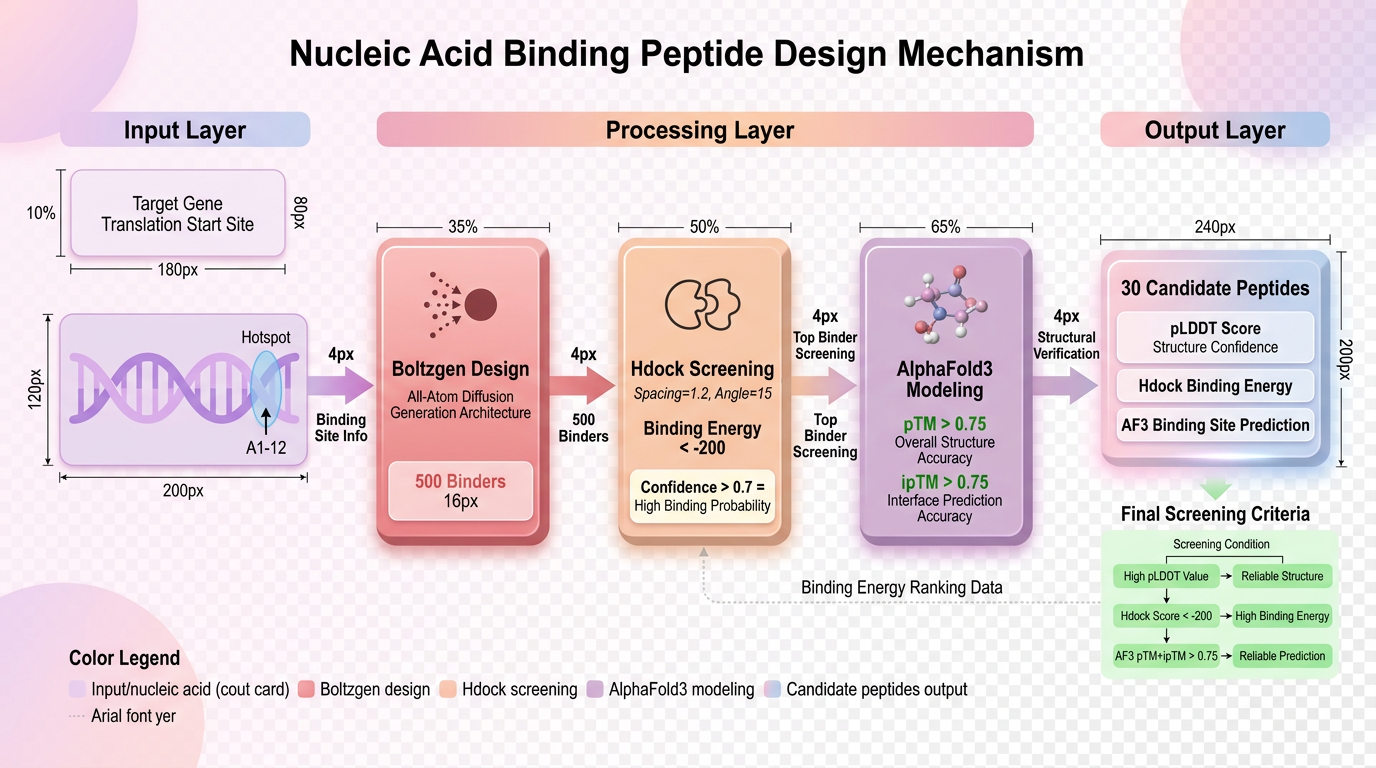
设计流程思路
第一步:锁定"靶心"------结合位点的精确确定
设计多肽的前提是了解靶点。在实际研发中,我们通常将目标基因的启始翻译位点及其周边区域作为重点干预对象。
利用AlphaFold3(AF3)强大的三维预测模型,可以精确建立目标核酸起始翻译位点的空间结构信息。通过确认特定序列(例如A1-12区域)的空间构象,为后续多肽的设计提供精确的互作结合界面的物理及化学限制条件。这就好比在制造钥匙前,先用3D扫描仪将锁芯的三维结构彻底摸透。
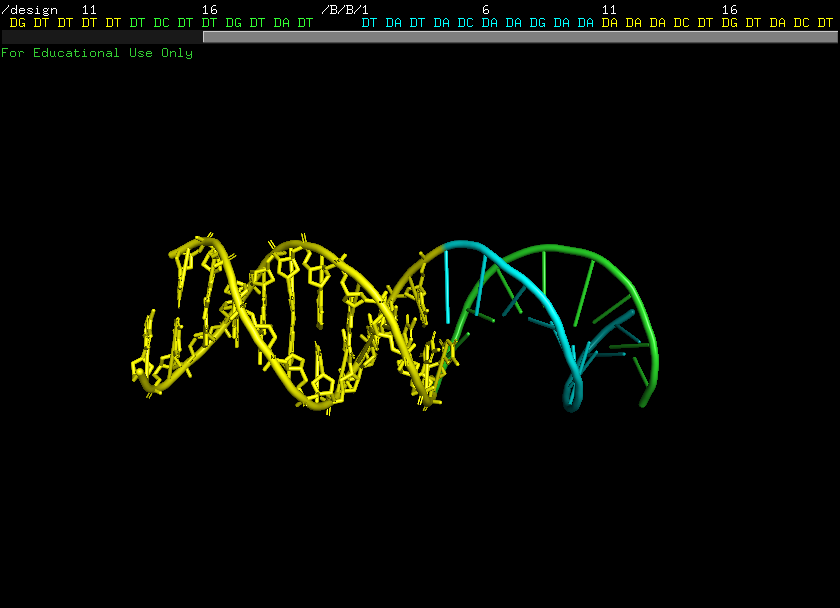
靶点确定示例
第二步:从无到有------基于Boltzgen的结合物生成
明确了结合位点,下一步就是"捏"出能够完美契合该位点的多肽。
这里应用到了生成式AI的尖端工具------Boltzgen 。这是一种利用全原子扩散生成架构的模型,专为生成具有高特异性和亲和力的蛋白质/多肽结构而生。
在科晶生物的技术实践中,会将结合物的长度设定为精准的30bp,并设定模型一次性从头生成500个完全不同的结合物(Binder)候选库。这种基于扩散模型的生成方式,不再依赖于天然蛋白质库的搜索,而是真正意义上的"从头设计(De Novo Design)",极大拓展了针对复杂核酸靶点的多肽化学空间。
第三步:大浪淘沙------HDOCK分子对接精筛
拥有了500个候选分子后,需要通过高效的算法进行筛选。
HDOCK 是一种融合了模板匹配和自由对接算法的高效工具。在这个阶段,为了获得高分辨率的对接结果,技术人员通常需要精细调节参数(如将Spacing和Angle分别精调为1.2和15)。
如何评判对接分数?
对接分数是一个预测指标(通常为负值,越小代表结合潜力越大)。根据HDOCK的官方模型测算,通常引入置信度公式进行换算。当对接分数达到 -200 左右时,其置信度(Confidence Score)即可达到 0.7 以上。
- > 0.7:两个分子结合的可能性极大,属于核心候选队列。
- 0.5 - 0.7:存在结合可能。
- < 0.5 :结合概率较低,直接淘汰。
通过这一轮算力筛选,可以将500个候选库迅速瘦身,仅保留结合能最低、结构最稳定的顶尖候选分子。
第四步:终极考验------AlphaFold3的一对一精细验证
最后,也是最严苛的一步,是将HDOCK筛选出的Top级数据,重新输入到AlphaFold3中进行高精度的复合体测算。
这里有两个非常核心的评价维度:pTM 和ipTM。
- pLDDT值与pTM(预测的TM分数): 用于评估多肽本身的局部预测可信度与整体折叠准确性。pLDDT值越高,说明模型预测的结构越接近真实世界的多肽三维结构。
- ipTM(界面预测TM分数): 这是评估核酸-蛋白质复合体的灵魂指标。它专门用来测算两个大分子之间"接触区域"的结构准确性。
在科晶生物的最终交付标准中,只有当 pTM + ipTM 的综合评分大于0.75 被认为是高置信度的对接效果。这意味着该多肽不仅自己折叠得好,且与目标核酸结合得异常紧密,后续进入湿实验(Wet Lab)合成及功能验证的成功率将大幅提升。
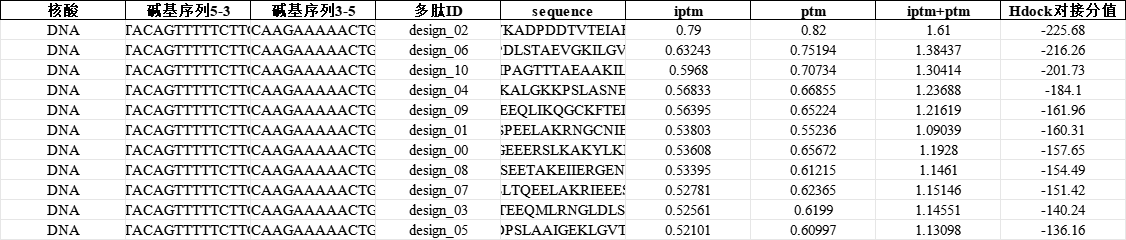
设计结果示例
总结:AI重构核酸药物研发范式
从明确靶点结构,到扩散模型生成,再到对接打分,最后由微观复合物模型盖棺定论。可以清晰地看到,如今的计算大分子设计已经完全脱离了"盲盒摸象"的阶段。
通过pLDDT值判断多肽三维结构的可信度、通过HDOCK测算物理结合能、通过AlphaFold3预测真实结合位点,科晶生物这套全链路的计算辅助设计方案,为科研工作者和创新药企提供了一个低成本、高效率的"雷达系统"。
在核酸药物(如ASO、siRNA的递送与调控)、基因编辑工具优化日益火热的今天,基于AI的"定制化核酸结合多肽"技术,必将成为加速生物医药产业跨越式发展的核心引擎。
参考文献:
- Hannes Stark, Felix Faltings, Jeremy Wohlwend, Gabriele Corso, Regina Barzilay, Tommi Jaakkola., et al. (2025). BoltzGen: Toward Universal Binder Design. bioRxiv,doi: 10.1101/2025.11.20.689494.
2.Yan Y , Tao H , He J ,et al.The HDOCK server for integrated protein--protein dockingJ.Nature Protocols, 2020, 15(Suppl 25):1-24.DOI:10.1038/s41596-020-0312-x.
3.Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493--500 (2024). https://doi.org/10.1038/s41586-024-07487-w.