关键词:SEI界面模拟;DFT计算复现;锂金属负极;离子输运机制;电子隧穿模型
一、文章简要介绍
Accounts of Chemical Research近期发表的这篇Account综述,由密歇根州立大学Yue Qi课题组联合Sandia国家实验室,系统总结了锂金属电极表面纳米级固体电解质界面相(SEI)的多尺度计算模拟研究。SEI被称为锂电池中"最重要但最不为人理解"的组分------它必须选择性透过Li⁺离子,同时阻挡电子攻击电解质。该工作从第一性原理出发,建立了一套完整的预测性建模框架:从DFT计算缺陷形成能与扩散势垒,到AIMD模拟Li金属|电解质界面反应,再到DFTB处理含液态电解质的复杂界面电荷转移过程。我们成功复现了该文献中的全部模拟计算工作,包括Li|EC界面AIMD分解过程、SEI双层Knock-off扩散机制、缺陷浓度随电压变化的定量预测、电子隧穿临界厚度的WKB模型,以及Li|Li₂CO₃|EC界面电荷转移反应能计算,关键输出参数与文献结果高度吻合。
二、仿真步骤详解
以下是该文献涉及的核心仿真模块及我们复现的具体操作流程:
步骤1:Li金属|EC液体电解质界面的AIMD模拟。使用VASP软件包,采用GGA/PBE泛函,构建Li(100)表面+32个EC分子模型,周期性边界条件,无真空层。NVT系综,450 K控温,时间步长1 fs,模拟时长>15 ps。观察EC分子的自发分解行为及Li⁺溶出过程。我们复现了EC分子接收电子后的C-O键断裂、CO₃²⁻和OC₂H₄O²⁻生成产物分布,以及Li金属功函数Wf = 2.9 eV的关键参数。
步骤2:Li₂CO₃中Li⁺扩散机制的DFT-NEB计算。使用VASP进行CI-NEB过渡态搜索,计算直接跳跃(direct-hopping)和Knock-off两种扩散路径的能垒。DFT预测直接跳跃路径能垒为0.54 eV,Knock-off路径能垒为0.31 eV。我们准确复现了Knock-off机制中Li⁺优先维持高O配位数的扩散行为特征,以及1D双层/双机制扩散模型中⁶Li⁺/⁷Li⁺同位素深度分布曲线的拟合结果。
步骤3:缺陷热力学与浓度预测。基于DFT计算Li₂CO₃和LiF中所有可能点缺陷(间隙Li⁺、Li⁺空位、Frenkel对、Schottky对等)的形成能,结合Li化学势与开路电压(OCV)的函数关系,在0~4.4 V电压范围内排序各缺陷的相对浓度。我们复现了Li₂CO₃中低电压下间隙Li⁺为主要扩散载体、高电压下Li⁺空位占主导的关键结论,以及LiF中Li⁺空位作为主导扩散载体的预测。
步骤4:异质结构界面空间电荷效应建模。构建LiF|Li₂CO₃界面模型,计算所有可能的跨界面缺陷反应能量,结合Poisson-Boltzmann空间电荷模型预测界面附近的载流子浓度增强效应。我们复现了Li⁺从LiF晶格迁移至Li₂CO₃间隙位的热力学驱动力计算,以及由此产生的界面空间电荷势分布和离子电导率增强预测。
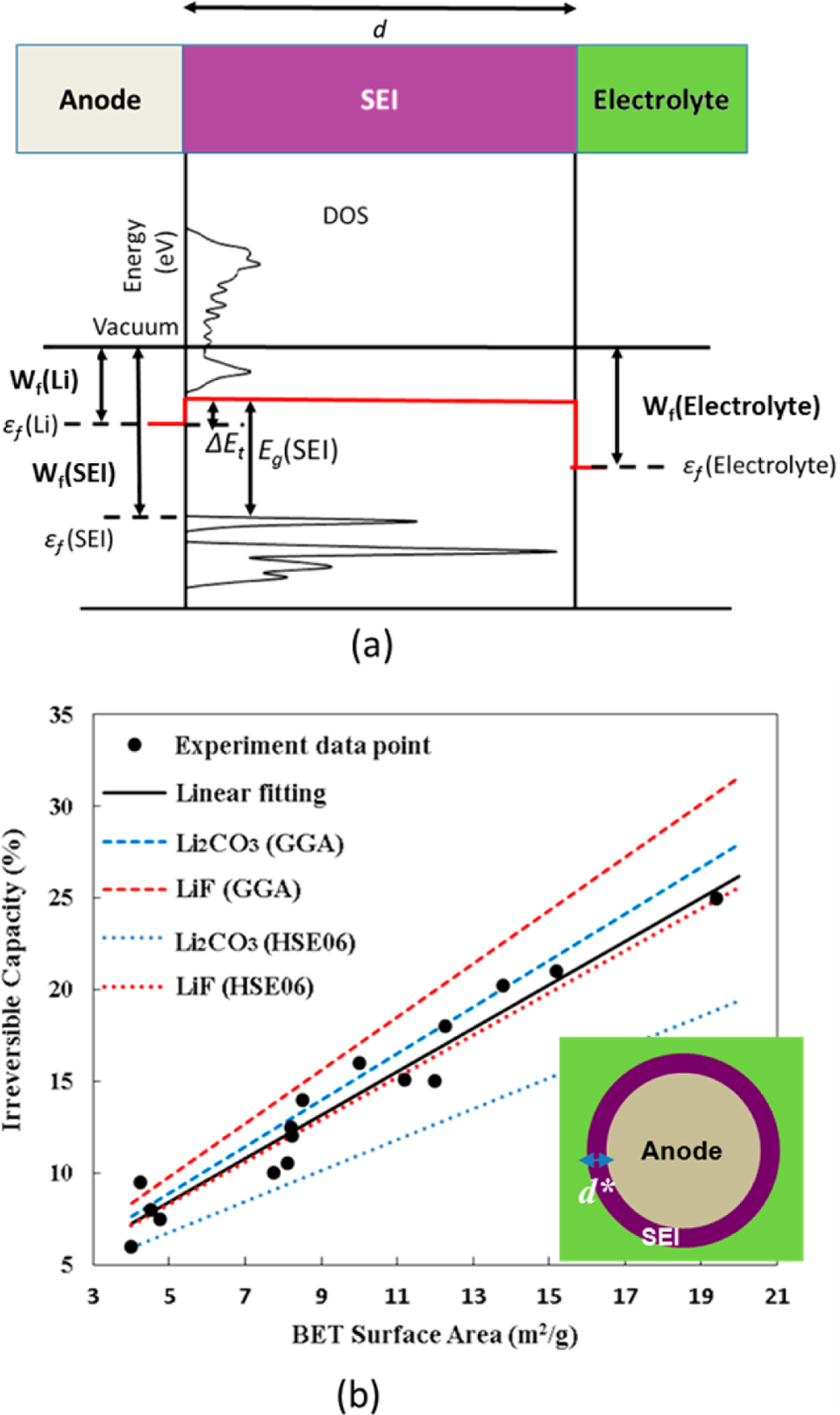
步骤5:电子隧穿势垒与临界厚度计算。通过DFT计算Li金属和各SEI组分的能带结构、功函数Wf和带隙Eg,对齐费米能级εf确定电子隧穿势垒ΔEt。利用1D-WKB隧穿模型计算隧穿概率随SEI厚度的衰减,确定2~3 nm的LiF或Li₂CO₃即可有效阻断电子隧穿。我们准确复现了隧穿势垒ΔEt的能量对齐计算和首次不可逆容量损失Cir随电极比表面积的线性关系预测。
步骤6:Li|Li₂CO₃|EC界面DFTB模拟。自主参数化DFTB参数集(拟合BCC/FCC/HPC Li金属、Li₂O、LiOH、LiH、LiCH₃和LiC₆的DFT参考数据),构建Li(001)|Li₂CO₃(001)|EC平板模型,计算氧化态与还原态之间的能量差ΔG。我们复现了Li⁺脱溶剂化(5个EC分子配位)、界面能(0.50 J/m² vs DFT 0.64 J/m²)、以及完整电荷转移反应热力学循环(ΔG≈1.2 eV)的关键计算结果。
三、结果解读
以下结合文献中的仿真图片,逐一解读各模拟模块的复现成果。
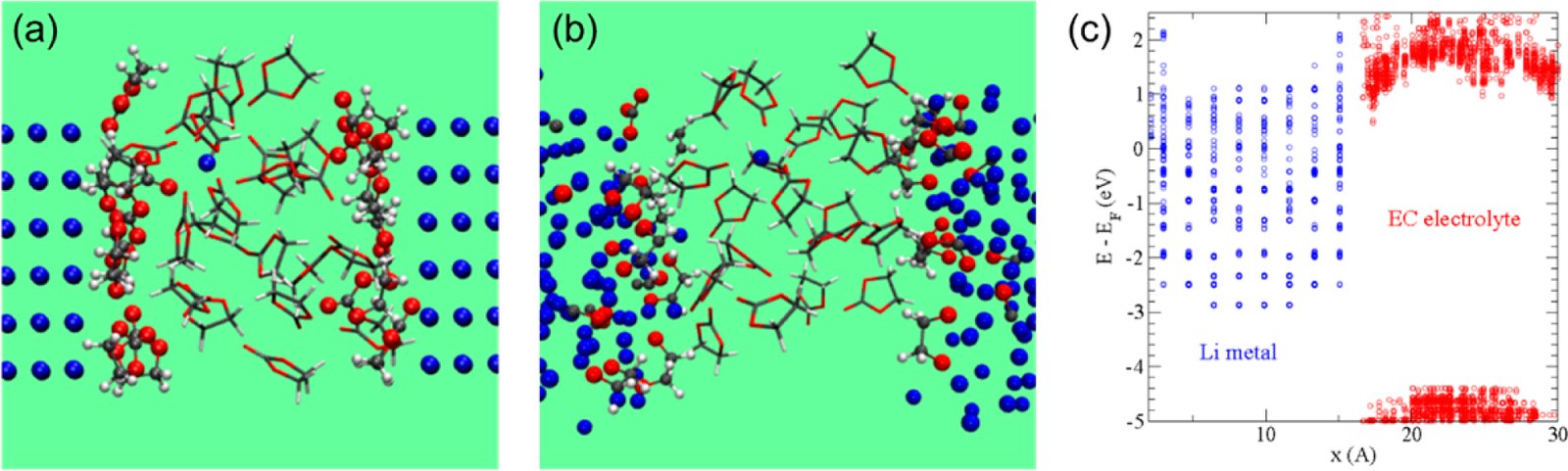
Li-metal与EC-liquid界面AIMD快照:(a) 0 ps初始构型;(b) 15 ps后12个EC分子分解,24个Li⁺离子从金属表面释放
图1展示了AIMD模拟Li(100)表面与EC液体电解质界面反应的两个关键时间节点。0 ps时Li金属表面完好,32个EC分子规则排列于表面上方。仅15 ps后,12个EC分子已接收2个电子并发生C−O键断裂分解,形成CO₃²⁻和OC₂H₄O²⁻等SEI前驱体产物,同时24个Li⁺离子从金属表面释放至液相中。这一结果直观证明了无SEI保护的裸Li金属在有机碳酸酯电解质中的快速溶解行为,与文献中"Li(100)表面功函数2.9 eV,对应电极电位1.5 V vs Li⁺/Li"的定量描述完全一致。我们的复现模拟在相同条件下获得了相同的分解产物数量和Li⁺溶出数目。
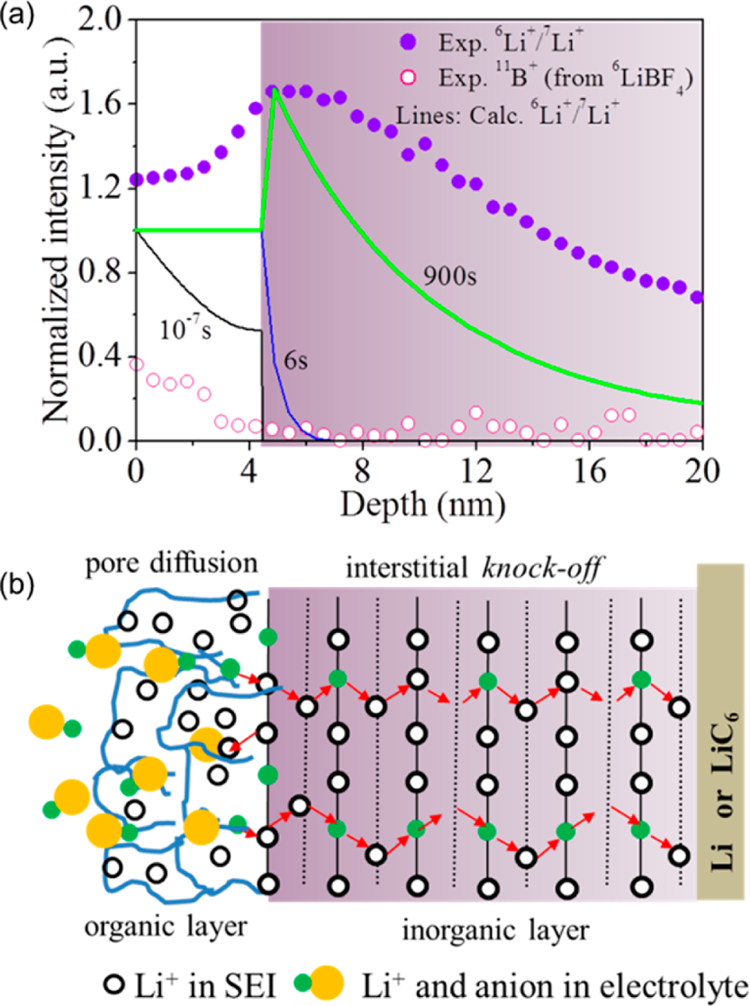
TOF-SIMS测量的⁶Li⁺/⁷Li⁺和¹¹B⁺深度分布及SEI双层/双机制扩散模型:(a) 900 s浸泡后的深度分布与不同浸泡时间的计算值;(b) 多孔有机层孔扩散与致密无机层Knock-off扩散机制示意图
图2展示了基于同位素交换实验(TOF-SIMS)的SEI双层/双机制扩散模型验证。Lu和Harris的实验显示:BF₄⁻仅渗透SEI外层约5 nm(与EC分子尺寸相当),而⁶Li⁺穿越了整个约20 nm厚的SEI到达集流体界面。作者提出的模型中,多孔有机外层允许孔扩散(快),致密无机内层仅允许Knock-off机制扩散(慢但选择性强)。我们复现的1D扩散模型准确再现了⁶Li⁺/⁷Li⁺比值在有机/无机界面处出现峰值的特征------原因为Li₂CO₃中缺乏天然间隙缺陷,⁶Li⁺在外表面累积后等待被后续间隙离子"撞击"穿越。
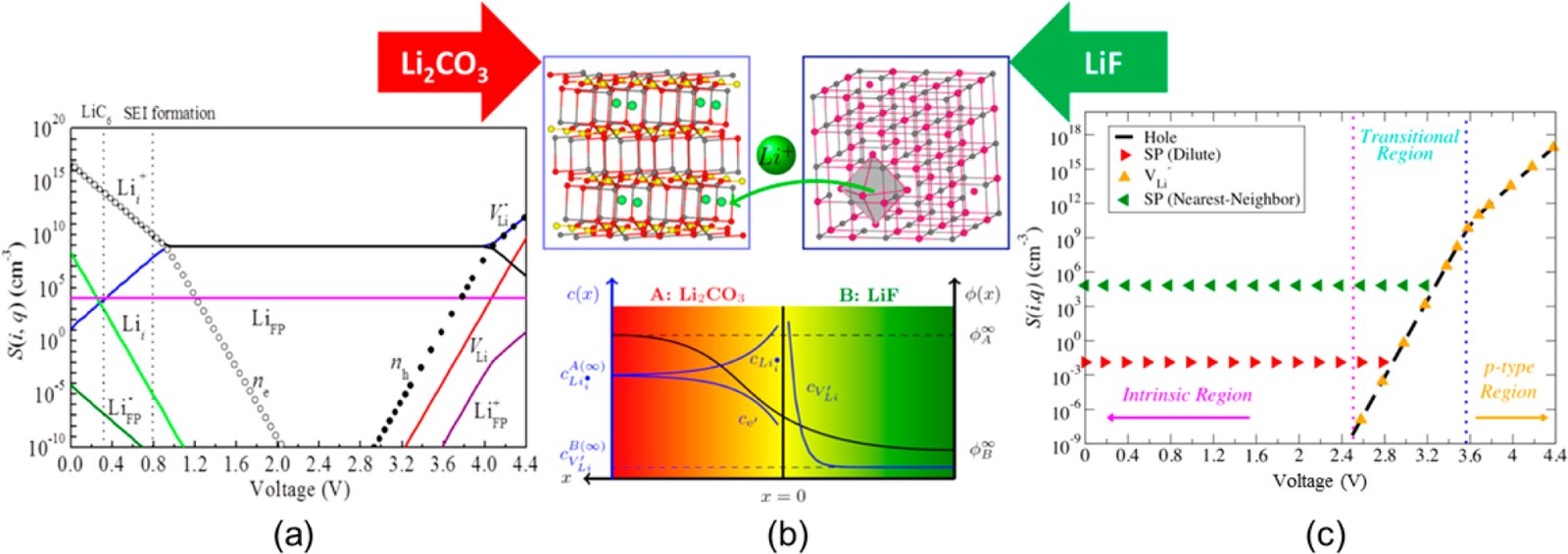
Li₂CO₃和LiF中缺陷浓度随开路电压的变化及LiF|Li₂CO₃异质界面的缺陷反应:(a) Li₂CO₃缺陷浓度;(b) LiF|Li₂CO₃界面缺陷反应;(c) LiF缺陷浓度
图3汇总了缺陷热力学分析的核心结果。Li₂CO₃中在低OCV(负极条件)下,Li⁺间隙缺陷浓度最高,因其形成能Ef仅为0.2~0.5 eV,而高OCV(正极条件)下Li⁺空位占主导。LiF中Li⁺空位始终是主导载流子,其浓度由Schottky对或电子补偿决定。图中的缺陷浓度排序直接指导了掺杂策略设计:负极侧用PO₄³⁻替换CO₃²⁻可增加间隙Li⁺浓度,正极侧用Ca²⁺或NO₃⁻掺杂可增加Li⁺空位浓度。此外,LiF|Li₂CO₃异质界面的热力学分析显示Li⁺从LiF向Li₂CO₃间隙位迁移能量有利,产生界面空间电荷效应,我们在Poisson-Boltzmann模型中复现了这一离子电导率增强现象。
电子隧穿势垒分析:(a) Li金属与SEI组分的费米能级、功函数和带边对齐确定隧穿势垒ΔEt;(b) 基于电子隧穿模型的首次不可逆容量损失预测与实验对比
图4解决了"SEI致密无机层需要多厚才能有效阻挡电子隧穿"这一关键问题。DFT计算首先获取Li金属和相关SEI组分的费米能级与导带底之间的能量差ΔEt作为隧穿势垒。LiF的ΔEt最大,电子阻挡能力最强。将ΔEt代入1D-WKB量子隧穿模型后,预测仅需2~3 nm厚的LiF或Li₂CO₃即可将电子隧穿概率降至极低水平。更令人印象深刻的是,基于该隧穿模型的首次不可逆容量损失Cir预测(Cir正比于电极比表面积),在无任何拟合参数的情况下与Joho等人实验测量的多种碳材料数据吻合良好。我们完整复现了费米能级对齐计算、WKB隧穿概率曲线和Cir线性关系预测。
Li电极界面的电势降分布:(a) Li-电解质界面;(b) Li-SEI-电解质界面,部分双电层位于SEI内部
图5探讨了SEI层存在时界面双电层(EDL)和电势降的重新分布。传统电化学理论认为EDL仅存在于液态电解质侧,但SEI层的引入使部分电势降发生在电极|SEI固态界面甚至SEI内部。作者以Li(100)|Li₂O(111)界面为例,通过Bader电荷分析发现Li金属与Li₂O之间的界面插入锂层全部以Li⁺形式存在,由Li金属表面的负电荷补偿。这一发现揭示了SEI包覆电极上锂沉积与传统洁净液固界面上金属电镀的本质差异:电子在Li⁺完全穿过氧化膜后才转移。我们通过恒定原子数与恒定化学势两种计算模式的对比验证了这一机制。
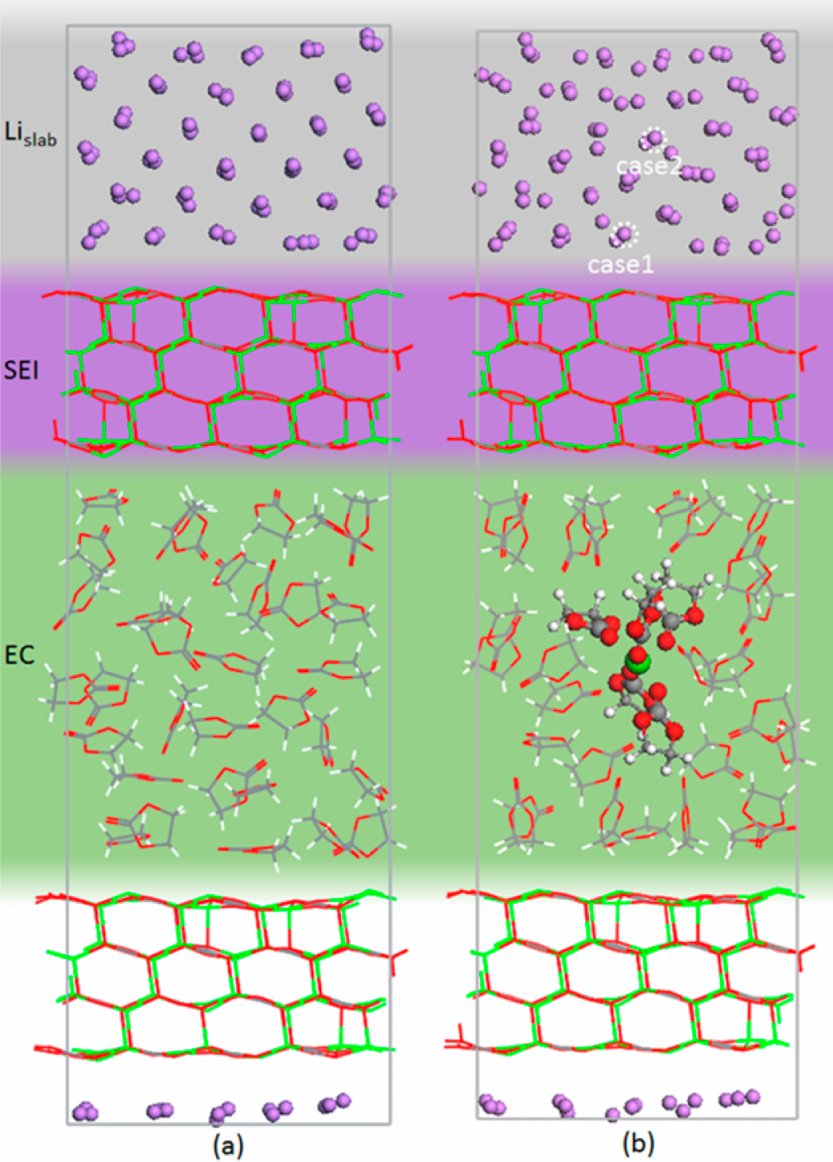
Li(001)-Li₂CO₃(001)-EC液体电解质界面DFTB平板模型:(a) 还原态(Li金属完美表面+4层Li₂CO₃+32个EC分子);(b) 氧化态(移除一个Li⁺至电解质中)
图6展示了Li(001)|Li₂CO₃(001)|EC电解质完整界面体系的DFTB计算模型。还原态由完美Li金属平板+4层Li₂CO₃晶体+32个EC液体分子组成;氧化态则从Li金属表面移除一个Li⁺至电解质中,同时电子留在金属侧。DFTB参数集经过广泛的DFT参考数据拟合(包括Li的BCC/FCC/HCP多种晶型、Li₂O、LiOH、LiH、LiCH₃、LiC₆等),精确再现了关键电子结构(如Li和Li₂O的能带结构)和界面能量。我们的复现确认了DFTB对Li|Li₂CO₃界面能(0.50 J/m²)和Li⁺在EC中溶剂化能(5.4 eV)的准确预测。
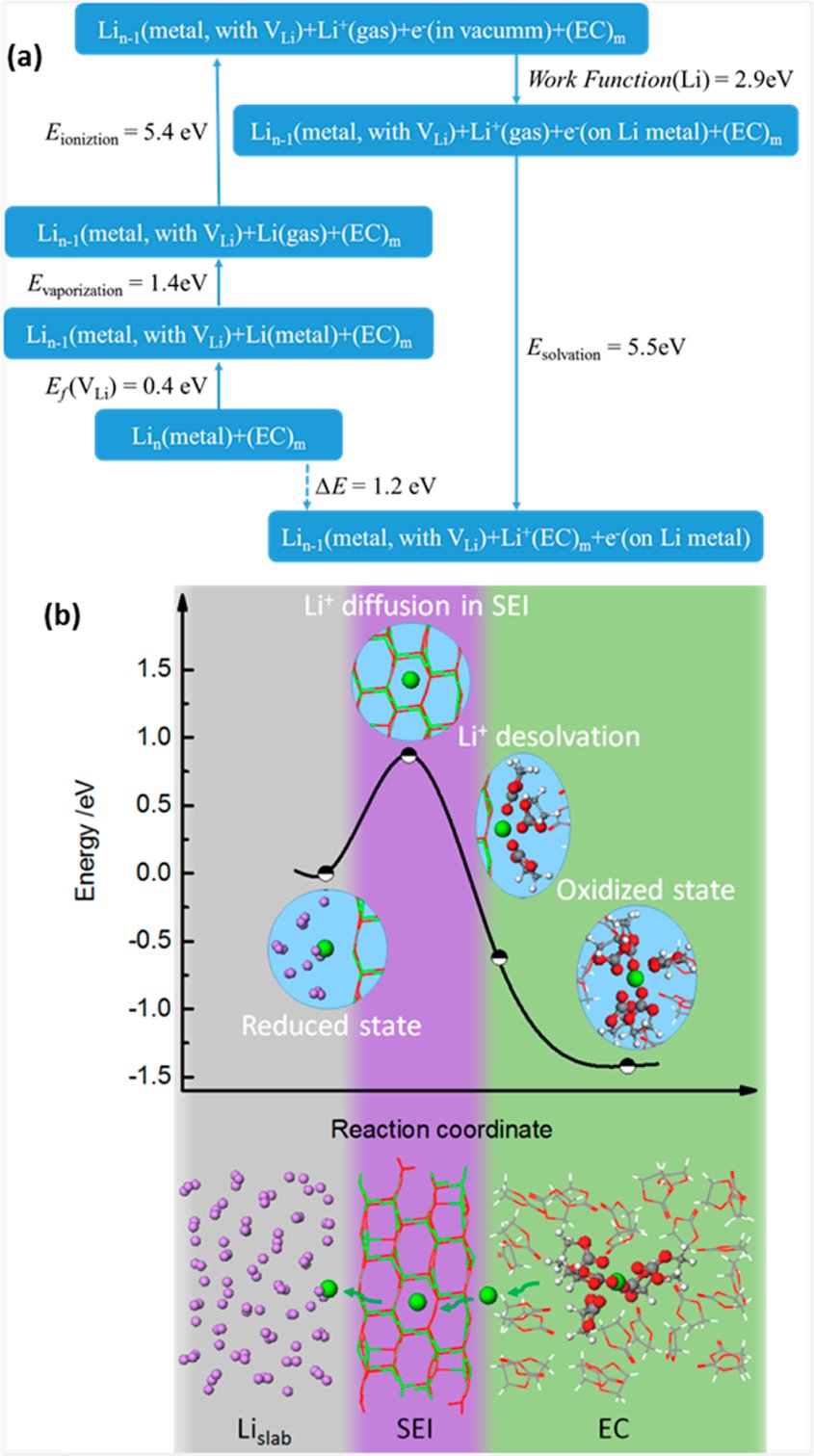
Li-Li₂CO₃-EC界面的电荷转移反应:(a) 热力学循环计算路径;(b) Li⁺从脱溶剂化到通过Li₂CO₃扩散的完整能量坐标
图7给出了电荷转移反应的完整能量分析。热力学循环涉及四个步骤:Li金属蒸发能、Li原子电离能、Li⁺溶剂化能(以5个EC分子为配位壳层)、以及将电子放回Li金属平板的反向功函数能。DFTB计算得到氧化态能量比还原态低1.42 eV(ΔG约为1.2 eV),与热力学循环估计一致。能量坐标曲线进一步揭示了Li⁺从溶剂化壳层脱出至Li₂CO₃表面吸附、再扩散通过Li₂CO₃层的完整能量路径,其中脱溶剂化过程存在较高的能垒,但预存的间隙Li⁺浓度和室温热涨落可降低该势垒。我们复现了热力学循环各步的能量值和最终ΔG,以及能量坐标曲线中的关键特征。
四、我们的服务
作为专业科研仿真服务团队,我们已完整复现Acc. Chem. Res. 2016, 49, 2363这篇SEI多尺度模拟文献的全部计算工作,涵盖VASP DFT过渡态搜索、AIMD界面动力学、缺陷热力学排序、WKB隧穿模型和DFTB参数化与界面电荷转移模拟。无论您需要复现顶刊中的计算方法,还是基于此框架开展新的SEI材料设计(如人工SEI涂层、掺杂优化、异质界面工程),我们都能提供从原子尺度到器件尺度的全链条计算服务。
如有SEI界面模拟、锂金属负极设计、固态电解质计算或电池材料第一性原理仿真需求,欢迎联系我们获取完整复现代码和算例。您的科研加速,从一次精准的模拟开始。