GenomeScope:基于未组装短读长序列的快速基因组分析 作者 :Vurture GW、Sedlazeck FJ、Nattestad M、Underwood CJ、Fang H、Gurtowski J、Schatz MC(2017) 期刊 :《生物信息学》 文献链接 :https://doi.org/10.1093/bioinformatics/btx153
bash
http://genomescope.org/ #在线版官网正文翻译
当前从头组装技术的发展,大多聚焦于结构相对简单的基因组。即便是人类基因组(杂合率仅约 0.1%,标准二倍体),其复杂度也远低于诸多其他物种,尤其是植物。如今基因组学研究正逐步转向解析高复杂度物种,例如菠萝、甘蔗、小麦:这类物种杂合度更高(菠萝杂合率>1%)、倍性复杂(甘蔗为八倍体)、基因组体量庞大(小麦基因组达 160 亿碱基对)。
测定新物种基因组时,首要目标之一就是解析基因组整体特征,包括基因组大小、重复序列占比、杂合率。这些指标不仅可用于研究基因组演化规律,还能为基因组组装的各项参数设置提供参考;同时也可作为独立质控依据,例如评估组装质量、在变异检测前预估基因组杂合位点数量。
我们开发了分析模型与开源软件 GenomeScope ,可直接利用未组装测序数据 推导基因组整体特征。该软件依托 k-mer 频数分布(常用工具 Jellyfish 统计得到),数秒内即可生成分析报告与可视化图谱,直观展示基因组各项属性。本研究通过模拟杂合基因组 、以及已有参考序列的微生物 / 真核生物菌株杂交数据集,验证了该方法的可靠性。目前 GenomeScope 也已应用于多个新物种研究,包括菠萝、梨、再生涡虫(Macrostomum lignano)与亚洲海鲈。
快速上手
使用 GenomeScope 前,需先统计k-mer 频数直方图 ,推荐工具 Jellyfish(下载地址:http://www.genome.umd.edu/jellyfish.html)。编译安装 Jellyfish 后,执行以下命令统计 k-mer:
jellyfish count -C -m 21 -s 1000000000 -t 10 *.fastq -o reads.jf参数说明
需根据服务器配置调整内存 (-s)和线程数 (-t);示例命令使用 10 线程、1GB 内存。
-m:设置 k-mer 长度。若测序深度偏低或测序错误率偏高,需酌情调整该值。- 绝大多数物种推荐使用 k=21:该长度既能保证大部分 k-mer 为非重复序列,同时对测序错误的容错性更强。
- 单倍体基因组远超 100 亿碱基对、或重复序列占比极高的物种,建议选用更长的 k-mer,以提升独有 k-mer 的数量。
关键要求
- 测序深度 :单倍体基因组测序深度至少达到25×,深度不足会导致模型拟合失败、无法收敛;
- 测序质量 :仅适用于低错误率数据(如 Illumina 测序)。若错误率达到 2%(平均每 50 个碱基出现 1 处错误),已超过常规 k-mer 长度(k=21);牛津纳米孔、PacBio 单分子测序数据(错误率 5%~15%)不支持使用;
- 必须添加参数
-C(统计标准 k-mer):测序读段同时包含 DNA 正、反链序列,该参数可保证统计结果准确。
导出 k-mer 频数直方图
bash
jellyfish histo -t 10 reads.jf > reads.histo同样可根据服务器性能调整线程数-t。得到直方图文件后,可使用网页版或命令行版运行 GenomeScope。
网页版运行
网页版操作简单、功能完整,访问地址:http://genomescope.org/
命令行版运行
依托 R 脚本genomescope.R分析,需确保Rscript已配置系统环境变量(也可修改脚本解释器路径):
Rscript genomescope.R 直方图文件 k-mer长度 读长 输出目录 [最大k-mer覆盖度] [详细输出]- 必选参数:直方图文件、k-mer 长度、测序读长、结果输出目录;
- 可选参数 :
最大k-mer覆盖度:过滤高拷贝 k-mer 的阈值,推荐设为 1000,可根据数据特征调整;详细输出:设为 1 时,输出模型拟合的详细日志。
分析完成后,图谱与基因组特征统计文本会自动保存至指定输出目录。
运行示例
可下载文中拟南芥 F1 代样本的直方图文件测试: https://raw.githubusercontent.com/schatzlab/genomescope/master/analysis/real_data/ara_F1_21.hist
执行命令:
/PATH/TO/Rscript /PATH/TO/genomescope.R ara_F1_21.hist 21 150 output程序 1 分钟内即可运行完毕,输出示例: Model converged het:0.0104 kcov:22.2 err:0.0035 model fit:0.446 len:151975724 结果解读:模型收敛,杂合率 1.04%,k-mer 平均覆盖度 22.2,测序错误率 0.35%,模型拟合度 0.446,预估基因组大小 151.9 Mb。
受模型随机运算影响,最终数值会存在小幅波动。
配套教程
Andrew Severin 编写的实操教程:http://gif.biotech.iastate.edu/genomescope
常见问题(FAQ)
1. 模型无法收敛,或分析结果与预期偏差极大?
最主要原因是测序深度不足,模型无法识别纯合 k-mer 对应的特征峰;表现为 k-mer 分布图无明显峰形、拟合曲线与实际数据偏差严重。 解决办法:
- 确认 Jellyfish 已开启标准 k-mer 统计(参数
-C); - 尝试将 k-mer 长度调低至 17 或 19;
- 以上操作均无效时,需补充测序数据、提升测序深度。
2. 单倍体基因组大小如何计算?为何要除以纯合 k-mer 覆盖度的 2 倍?
首先区分变量定义:文献中用λ 、代码中用kcov,二者指代同一数值(杂合 k-mer 的平均覆盖度)。 模型会识别 4 个特征峰,中心位置依次为 λ、2λ、3λ、4λ,分别对应:独有杂合序列、独有纯合序列、重复杂合序列、重复纯合序列的平均覆盖度。
单倍体基因组计算公式:(剔除测序错误后的总 k-mer 数量) ÷ 纯合 k-mer 平均覆盖度(2λ)。
举例说明:人类基因组单倍体约 30 亿碱基对,二倍体约 60 亿碱基对。若总测序数据量为 300 Gb,相当于父、母本单倍体各获得 50× 测序深度。由于人类杂合度极低,分布图主峰对应 100× 覆盖度。此时模型判定:杂合 k-mer 覆盖度 λ=50×,纯合 k-mer 覆盖度 2λ=100×。总测序量 ÷ 纯合覆盖度(100×),即可算出单倍体基因组大小为 3 Gb。 若两套单倍体序列长度差异较大,最终输出结果为两者平均值。
3. GenomeScope 能否分析多倍体基因组、估算倍性?
仅支持标准二倍体基因组 ,暂不兼容多倍体。理论上可通过拓展模型识别更多峰形以适配多倍体,但目前软件未实现。 同时也不支持非整倍体样本(如肿瘤细胞、性染色体数目异常样本):此类样本输出的杂合率,会混淆单拷贝区域、双拷贝区域及染色体杂合位点的占比。
4. 传统 k-mer 法预估基因组 869~919 Mb,GenomeScope 结果仅 650 Mb,偏差超 200 Mb?
核心原因是高拷贝 k-mer 过滤阈值 :真实数据中超高拷贝 k-mer 多为噬菌体、细胞器 DNA 等污染序列,属于干扰信号。 软件默认过滤覆盖度>1000的 k-mer,这会直接拉低基因组预估大小。 示例:默认阈值下结果为 649 Mb;将阈值上调至 10000,结果变为 697 Mb。
解决办法:
- Jellyfish 默认会截断高覆盖度统计,若需纳入超高拷贝 k-mer,重新运行 Jellyfish 生成完整直方图;
- 上调 GenomeScope 的最大 k-mer 覆盖度阈值(可设为 10 万、100 万);
注意:阈值越高,结果越易受污染序列干扰,需根据数据情况权衡。详细参考原文补充材料(拟南芥高拷贝 k-mer 分析章节)。
5. 两个近缘鱼类样本单独分析杂合率分别为 0.103%、0.113%,合并读段后杂合率降至 0.056%?
两个样本合并后,数据等效为四倍体 ,而 GenomeScope 仅适配二倍体模型,因此分析结果失真。 混合数据会产生新的杂合 k-mer 峰,但软件会误将其判定为测序错误。目前团队正在开发多倍体适配模型。 验证方案:将读段比对至基因组组装序列,可检测出两种等位基因频率的变异位点。 多倍体模拟代码仓库:https://github.com/schatzlab/genomescope/tree/master/analysis/genomesim/polyploid
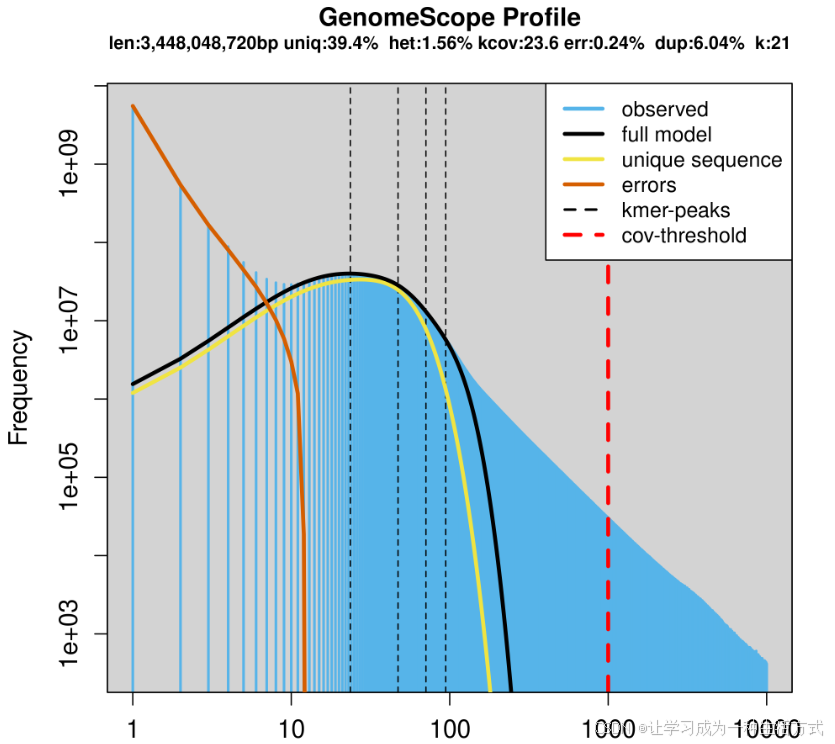
6. 图谱中各项指标含义?
完整定义见原文补充方法,简要释义:
- len:预估基因组总长度
- uniq:基因组中非重复序列(独有序列)占比
- het:整体杂合率
- kcov:杂合位点对应的 k-mer 平均覆盖度(真实数据存在离散分布,峰顶不会与 kcov 标线重合)
- err:测序读段的错误率
- dup:读段平均重复率
相关
大型基因组变异位点 VCF 文件:http://labshare.cshl.edu/shares/schatzlab/www-data/genomescope/vcf/
参考文献
Vurture GW, Sedlazeck FJ, Nattestad M, et al. (2017) Bioinformatics. 文献 DOI:https://doi.org/10.1093/bioinformatics/btx153