Chromosome-scale genomes of water lilies provide evolutionary insights into the aquatic adaptation of angiosperms
睡莲染色体水平基因组为揭示被子植物水生适应演化提供依据

核心要点
- 完成三种睡莲基部物种染色体水平基因组组装
- 睡莲目古老全基因组加倍事件推动基因组革新与类群分化
- 类群特异性基因增减是水生关键表型演化转变的分子基础
- 代谢通路重编程助力物种形成水生环境生理适应
摘要
被子植物的起源与分化历程,尤其是其祖先生态位演化问题,一直是学界长期未解的难题。目前相关演化假说层出不穷,但被子植物多次向水生环境趋同演化的基因组驱动机制仍尚不明确。
本研究组装获得水盾草、长叶宝莲芡、莼菜 三种基部睡莲植物的染色体水平基因组,并结合已发表睡莲目物种基因组开展比较基因组学分析。结果表明,睡莲类植物的水生适应性演化,由古老全基因组加倍事件与核心基因功能模块重塑共同驱动;具体表现为光合作用、胁迫响应相关基因家族发生扩张,核苷酸结合富亮氨酸重复序列(NLR)免疫受体基因家族出现收缩,同时萜类 - 鲨烯等逆境相关次生代谢通路发生代谢重编程。
上述基因组层面的变异特征,有效提升了物种在水生环境中的能量利用效率、免疫调控能力与资源适配能力。本研究证实了早期睡莲植物次生水生适应演化的重要作用,其特有的水生性状特征,均可通过长期适应沉水生存环境所产生的一系列演化改变得到合理解释。
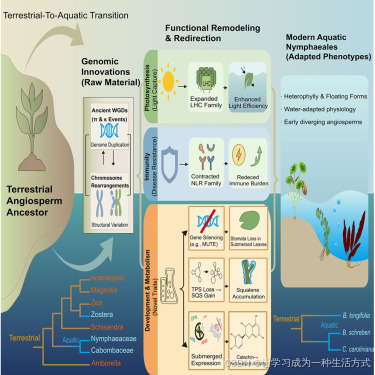
引言
被子植物的起源与早期演化,始终是植物学与进化生物学领域尚未解决的核心科学问题。尽管相关研究层出不穷,但学界依旧存在诸多争议。尤其针对早期被子植物的祖先生态位,目前主流假说观点截然相反。
其中一种观点认为,被子植物祖先为喜阴陆生林下木本植物,偏好温暖湿润、弱光照生长环境,依靠保守且节能的生存策略完成开花与传粉过程,无油樟以及木兰目等早期分化类群常被视作典型代表。另一种假说则提出,最原始的被子植物祖先可能为水生或半水生植物,水生环境带来的独特选择压力,推动其逐步演化形成水生生活习性。该观点的主要依据,多来自睡莲目这类早期分化且高度特化的水生被子植物类群。
但目前学界仍无法确定,这类水生性状究竟是被子植物原始祖先固有特征,还是陆生祖先在演化过程中二次适应水生环境形成的衍生性状。睡莲目隶属于被子植物基部 ANA 类群,是其中最具代表性的水生演化支,可为探究被子植物早期辐射分化过程中水生适应性演化历程提供重要研究材料,但其并不能直接等同于被子植物祖先类群。
这场学术争论并非单纯的理论探讨,更直接关乎两大核心进化命题:开花性状如何起源、被子植物为何能够实现全球性繁盛。祖先不同的生存生态位,会使植物在资源分配模式、生殖系统构建、生长与防御权衡策略上走向完全不同的演化方向,进而长期影响物种类群分化与形态建成规律。因此厘清被子植物原始生存生态位,能够进一步完善人们对植物性状创新演化机制及其宏观进化效应的认知。
水生环境与陆地环境存在截然不同的理化及生物限制条件。水体光照衰减速度更快、光谱组成发生改变,使沉水植物面临完全迥异的光照环境;水体中二氧化碳、氧气等气体扩散效率大幅降低,深水区域气体交换受阻问题尤为突出。同时,水生环境水流冲击、水体浮力、养分分布均存在极强时空异质性,水生植物还需适应多变的水流、水温、盐度环境,以及与陆生植物截然不同的微生物群落和病原菌胁迫。
为在水生环境中高效完成光能捕获、气体交换并维持机体稳态,植物需要对叶片、根系、维管组织、表皮结构进行大规模协同改造,同时重新调整代谢模式与免疫防御体系,重塑抗氧化系统及防御类次生代谢通路。大叶藻等海草的基因组研究已证实,植物向水生环境演化的过程中,会形成特征鲜明的适应性基因组变异特征;全基因组加倍事件及其后续基因层面的变异革新,更是助力植物实现适应性辐射演化、开拓全新生态位的重要基础。
基于以上研究背景,本研究聚焦核心进化问题:大规模基因组变异事件(全基因组加倍、染色体重排等),如何与基因家族扩张收缩、调控网络重塑、代谢通路改写等精细功能变异协同作用,共同驱动植物适应水生生态环境?
全基因组加倍是推动被子植物宏观性状革新的核心驱动力,该事件可一次性复制全部编码基因,为物种性状演化提供充足的遗传原材料。但基因复制本身无法决定演化走向,多倍化后的基因分化、剂量平衡约束、顺式调控元件重组、染色体结构变异等后续演化过程,直接决定复制基因的留存方式,以及基因功能发生增强、分化或丢失。
由此可见,多倍化能否产生适应性演化优势,完全取决于后续的功能重塑过程,只有将基因组潜在遗传优势转化为适应性表型,才能真正推动物种演化。多次全基因组加倍能够积累充足的演化潜能,而这种潜能能否发挥作用,则由基因留存模式与功能模块协同重编程共同决定。
想要验证上述演化规律,需选取系统发育地位原始、同时具备完整水生生活史特征 的植物类群开展研究,睡莲目恰好满足这一研究条件。作为被子植物早期分化演化支,睡莲目植物具备典型水生性状与多样化生存策略,是解析植物水生适应分子机制的天然研究体系。
此前已有团队完成首个睡莲基因组测序研究,但受限于样本数量,无法开展深入的比较基因组分析。本研究选取 3 种代表性睡莲目植物开展研究,包含两种全沉水植物:长叶宝莲芡、水盾草,以及浮叶植物莼菜,完成三者染色体水平基因组组装,并搭建多组学分析研究体系。
依托上述研究数据,本研究系统解析了早期水生被子植物类群多倍化事件的发生时间与系统发育分布规律,探究光能利用、免疫调控、次生代谢等核心功能模块是否存在协同定向演化特征,阐明基因组变异与功能重塑协同作用助力植物适应水生环境的分子机制。
其中,水盾草科与睡莲科互为姊妹类群,进化地位特殊;该科仅现存莼菜属与水盾草属两个属,物种遗传多样性精简,是研究植物性状演化与环境适应性的理想材料。
本研究通过构建睡莲目精准系统发育树,结合比较基因组与基因家族分析,重点解答三大科学问题:
- 多次多倍化事件在早期水生被子植物类群中如何分布,又为物种后续环境适应性演化提供了哪些遗传基础?
- 睡莲目植物体内,维系水生生存必需的光能捕获、免疫防御、特化代谢等核心功能模块,是否存在模块化趋同演化或定向演化规律?
- 早期分化水生被子植物的基因组变异与功能演化特征,能够为还原被子植物早期水生适应演化历程提供哪些理论依据?
本研究不仅阐明了睡莲植物水生适应性演化的遗传基础,也为揭示早期分化被子植物水生适应的基因组与功能分子机制,以及植物多次生境转换演化研究提供全新思路。
结果
两种沉水睡莲与一种浮叶睡莲的染色体水平参考基因组
本研究成功组装得到 3 个高质量染色体水平基因组,涵盖睡莲目不同水生生活型物种,分别为浮叶植物莼菜,以及全沉水植物水盾草、长叶宝莲芡。 三者基因组大小差异显著:莼菜基因组约 1.08 Gb,水盾草约 3.20 Gb,长叶宝莲芡约 0.67 Gb。基因组组装连续性优异,三者 Contig N50 依次为 16.0 Mb、47.6 Mb、18.1 Mb,Scaffold N50 分别为 28.7 Mb、59.7 Mb、37.8 Mb。 BUSCO 完整性评估结果显示,三份基因组完整度均高于 95%,组装质量可靠,可满足后续比较基因组学分析需求。基因组重复序列占比介于 59%~64% 之间,基因组大小差异主要由转座子扩增程度不同导致。
与核基因组存在明显的动态演化差异,三种睡莲植物的叶绿体基因组与线粒体基因组,在序列长度和基因组成上高度保守。三者叶绿体基因组长度分别为 159 kb、160 kb、159 kb;线粒体基因组长度依次为 396 kb、620 kb、599 kb。
核基因组结构多样且变异活跃,细胞器基因组却高度保守,二者形成鲜明对比,也体现出植物双重演化策略:核基因组通过不断结构重组积累变异,支撑物种适应不同水生环境;而叶绿体、线粒体等细胞器基因组序列高度稳定,保障植物光能转化、基础物质代谢等核心生命活动稳定运转,以此适配水生生存环境。
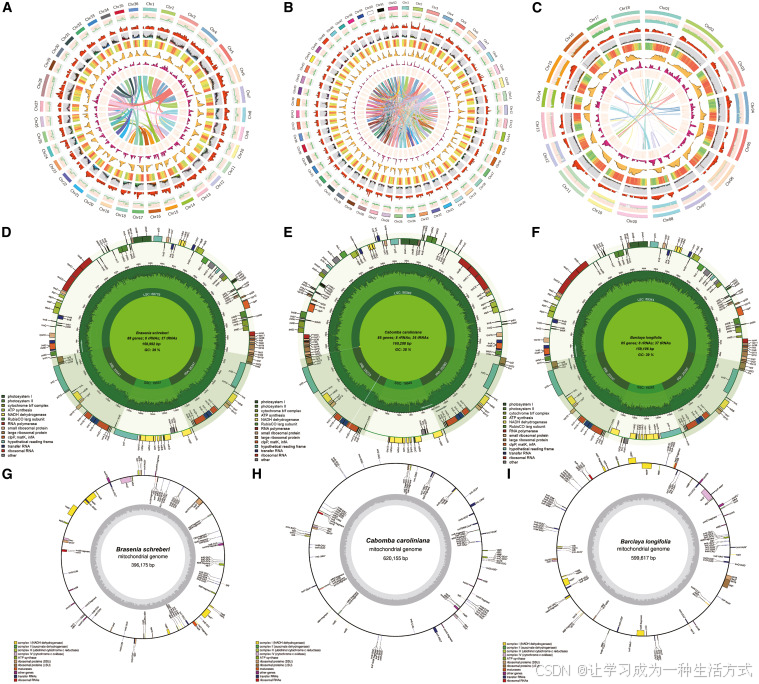
图 1 三种睡莲的核基因组、叶绿体基因组与线粒体基因组特征
(A~C)依次为莼菜、水盾草、长叶宝莲芡的圈图分析。由外圈至内圈依次展示:染色体序列、GC 含量、长末端重复序列组装完整性指数(所有 LTR 反转座子中完整 LTR 反转座子占比)、基因数量分布、重复序列占比、已注释全长 LTR 占比、完整 LTR 反转座子占比、Copia 型转座子分布以及染色体共线性关系。
(D~F)三种植物叶绿体基因组图谱,三者基因数量与基因类型高度相近,序列保守性极强。 (G~I)三种植物线粒体基因组环状图谱,展示整体基因组结构与基因排布特征。 整体而言,三种植物质体基因组高度保守,而核基因组在重复序列含量与 LTR 组成上存在明显差异。
古多倍化事件:水生适应性演化的重要驱动力
通过同义替换率(Ks)分布与共线性区块分析发现,睡莲目植物经历了复杂的多倍化演化历程。以往研究在睡莲属中鉴定出发生于约 1.2 亿年前的π 型全基因组加倍事件 ,但该加倍事件是否在睡莲科与水盾草科中共同发生,此前尚无定论。本研究借助新组装基因组明确解答了这一问题。
莼菜、水盾草、长叶宝莲芡与睡莲属物种的 Ks 分布曲线均在 Ks≈1 处出现显著峰值;睡莲属与莼菜的种间 Ks 比对结果显示,物种分化峰值出现时间略早于 π 全基因组加倍事件峰值,二者存在部分重叠。为提升推算精度,本研究结合类群特异性碱基替换速率完成 Ks 速率校正。
基因组共线性散点图结果进一步佐证上述结论:睡莲属与莼菜的同源共线性区块比例为 1:6,而非 2:12,有力证实π 全基因组加倍事件发生在两个类群分化之前 ,为睡莲科与水盾草科的共有古老加倍事件(图 2C)。
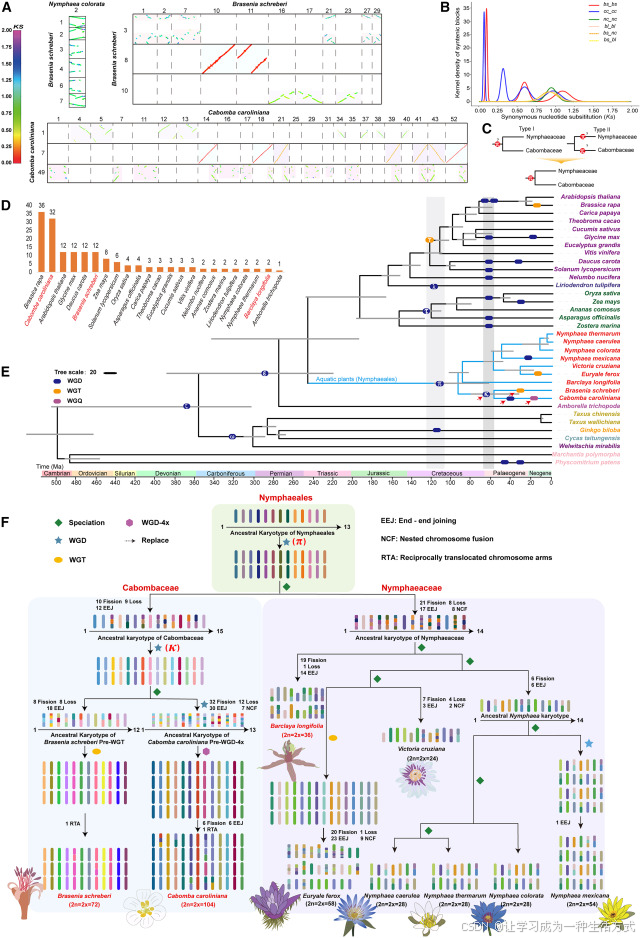
图 2 睡莲目物种基因组加倍事件与系统发育分析
(A)物种内部及物种间同源基因组比对。红花睡莲 2 号染色体与莼菜 1、2、3、4、6、7 号染色体存在大范围同源区段,二者同源比例为 1:6。莼菜基因组自身比对结果证实其经历三次基因组加倍:近期全基因组加倍(红色区块,1:2)、较古老加倍事件(绿色区块,1:3)、远古全基因组三倍化事件(浅绿区块,1:6)。水盾草共发生四次加倍:近期全基因组四倍化(红色,1:3)、后续全基因组加倍(橙色,1:4)、又一次全基因组加倍(绿色,1:8)以及最古老全基因组加倍(浅绿,1:16)。
(B)莼菜、水盾草、长叶宝莲芡、红花睡莲同义替换率(Ks)分布。种内比对峰值对应各物种特有全基因组加倍事件;种间 Ks 比对证实睡莲目物种均经历共同的 π 型全基因组加倍,图中展示经演化速率校正后的 Ks 数值。
(C)联合 Ks 分析与同源区块比对,明确证实睡莲目普遍存在 π 型古加倍事件。
(D)20 个物种倍性对比结果,显示水盾草相较于长叶宝莲芡发生了更多次基因组加倍。
(E)基于 34 个物种构建系统发育树并估算分化时间,证实水盾草科与睡莲科均发生多次基因组加倍,揭示睡莲目早期复杂的演化历程。
(F)核型演化推演显示,睡莲目祖先基因组含有 15 条染色体。经过结构重排与物种分化,逐步形成水盾草科与睡莲科各自的祖先核型。 本研究追溯了从睡莲目祖先类群,到各科祖先类群,再到现存物种的染色体演化路径,涵盖水盾草、莼菜、芡实、墨西哥睡莲、热带睡莲、克鲁兹王莲、红花睡莲、蓝睡莲、长叶宝莲芡等物种。
此外,本研究鉴定出水盾草科特有分支特异性全基因组加倍事件,命名为κ 事件 ,其 Ks 峰值集中在 0.5~0.75 区间。水盾草的基因组加倍历程更为复杂:在 κ 事件之后,该物种又先后发生两次物种特异性加倍、一次二倍化重建,再叠加高阶多倍化,最终推演倍性高达 32 倍。 该倍性为目前已报道早期分化被子植物中的最高水平,仅次于白菜的 36 倍,体现出水盾草为适应全沉水生活史发生了剧烈的基因组结构重塑。 与之不同,莼菜仅发生分支特异性全基因组三倍化,推演倍性为 12 倍;而长叶宝莲芡仅保留古老 π 型加倍事件,即便生存环境与水盾草相近,其加倍模式仍与睡莲属物种保持一致。
上述结果表明,睡莲目不同支系拥有截然不同的多倍化演化历史,各类群演化出差异化基因组策略以适应水生环境。水盾草科频繁发生支系特有加倍事件,而睡莲科仅保留共同的 π 古加倍,二者差异尤为显著。但这类加倍事件如何直接驱动后续基因家族扩张与功能创新,仍需进一步解析复制基因的起源与演化规律。
结合共线性分析与最大简约法重构推演,睡莲目祖先核型含有 13 条染色体。经过染色体末端融合、嵌套融合、染色体臂相互易位以及多次染色体断裂与丢失等结构变异,逐步分化形成水盾草科与睡莲科各自的祖先核型。本研究清晰还原了从睡莲目祖先到现存物种的染色体演化路径,厘清了各演化节点上的染色体逐步变异过程。
相较睡莲科,水盾草科核型演化活跃度显著更高:水盾草祖先核型历经 32 次染色体断裂、12 条染色体丢失、30 次末端融合与 7 次嵌套融合;而睡莲属祖先核型仅发生 6 次断裂与 6 次末端融合。 在经历共有 π 加倍与水盾草科特有 κ 加倍后,水盾草科还相继发生三倍化与四倍化事件。这类晚期多倍化伴随的基因组结构变异相对有限,大概率是因为加倍后演化时长不足,结构重排未能充分积累。 该差异也提示,多倍化发生初期往往伴随剧烈基因组结构变异;解读不同加倍事件的演化差异时需谨慎区分,这类差异更多源于加倍事件发生年代不同,而非物种核型重塑能力存在本质区别。
睡莲目水生适应机制:光感知与胁迫响应基因家族扩张
为揭示睡莲目植物向水生生活型转变的分子基础,本研究选取代表性被子植物开展比较基因组分析,系统探究同源基因簇的扩张收缩规律、功能富集特征、蛋白结构域组成,同时解析核心基因家族的拷贝数变异、表达模式与系统发育关系。 全基因组同源基因簇聚类及类群比对结果显示,睡莲目演化主干上存在大量显著扩张的基因家族,既存在目级共同祖先起源的家族扩张,也包含各分支独立演化形成的家族扩张。 韦恩图分析筛选出睡莲目特有及显著富集的同源基因簇,功能集中于光信号响应、叶片与气孔发育、各类逆境应答通路。 基因本体功能富集结果显示,扩张基因簇显著富集于光刺激应答、光形态建成调控、昼夜节律调控、生长素运输、气孔复合体发育调控 等通路;同时大量富集水分胁迫、盐胁迫、低氧胁迫应答 等水生适应核心通路。 睡莲目特有基因还显著参与防御应答、黏液代谢、生物胁迫响应、叶片衰老调控等过程,说明水生适应性演化不仅需要耐受非生物逆境,还需应对各类生物胁迫压力。
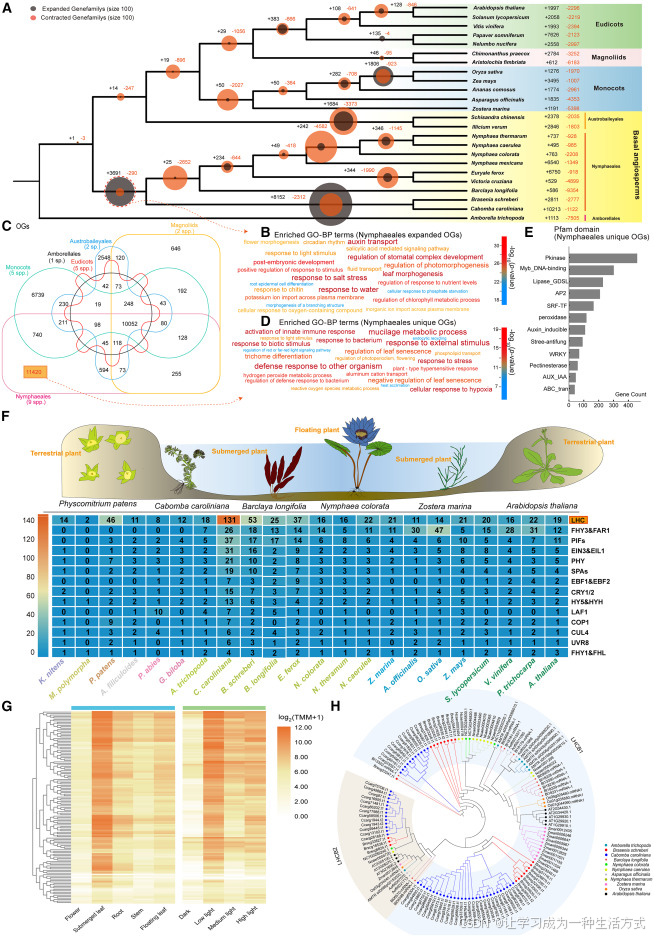
图 3 睡莲目植物水生适应相关核心基因扩张
(A) 24 种代表性植物(真双子叶、木兰类、单子叶及 ANA 基部被子植物)的基因家族扩张与收缩情况。利用 OrthoFinder 筛选同源基因簇,结合 CAFE 软件预测基因家族扩张与收缩事件。圆圈大小与扩张 / 收缩同源基因簇数量成正比;黑色数字代表扩张基因簇数目,橙色数字代表收缩基因簇数目。 (B) 睡莲目演化分支处扩张同源基因簇的 GO 生物学过程富集分析,其功能大多与环境适应相关。 (C) 相较于真双子叶植物、单子叶植物、木兰类植物、木兰藤目及无油樟目植物,睡莲目特有同源基因簇统计结果(附图 10)。 (D) 睡莲目特有同源基因簇的功能富集分析。 (E) 睡莲目特有同源基因簇的 Pfam 蛋白结构域注释结果。 (F) 选取轮藻植物、苔类、藓类、木贼类、无油樟目、睡莲目、单子叶植物与真双子叶植物共 22 种代表物种,开展光响应相关基因家族分析。结果显示水盾草体内 ** 捕光复合物(LHC)** 基因数量显著更高。 (G) LHC 基因在 5 种组织(花、沉水叶、浮水叶、根、茎)及 4 种光照条件(强光 12000 勒克斯、中光 2000 勒克斯、弱光 500 勒克斯、黑暗)下的表达热图。 (H) 11 个物种 LHCB1/2 基因的系统发育树,证实该基因家族在水盾草中发生显著扩张。
Pfam 结构域注释进一步阐明了睡莲目特有同源基因簇的功能特征(图 3E)。发生扩张的蛋白结构域包括富亮氨酸重复受体及类受体蛋白(LRR)、蛋白激酶(Pkinase)、Myb DNA 结合结构域、AP2 转录因子、GDSL 脂肪酶、C3HC4 环指蛋白、过氧化物酶与细胞色素 P450。 上述扩张结构域说明睡莲目植物的信号感知、转录调控、细胞壁与胶质物质代谢、病原菌防御能力均得到提升,与 GO 功能富集分析得出的结果一致。
在具体基因家族层面(图 3F),睡莲目多个参与光能捕获与光信号感应的基因家族出现明显扩张,包含捕光复合物基因 LHC、FHY3/FAR1、光敏色素互作因子 PIF、EIN3/EIL1、光敏色素 PHY、隐花色素 CRY、SPA、COP1 及 CUL4 等。 其中LHC 基因家族 拷贝数增幅最为显著,水盾草拥有多达 131 个 LHC 基因,在所有研究物种中数量最高,且 LHCB1、LHCB2 两个亚家族大规模扩张(图 3H;附表 8、附表 9)。 这类基因编码定位于类囊体膜上的光系统 Ⅱ 捕光蛋白,既能够捕获并传递光能,还可参与非光化学淬灭等光保护生理过程。 转录组数据表明,这类扩张基因在受光组织(如沉水叶)以及弱光环境下高量表达(图 3G)。 这表明该基因家族不仅发生拷贝数扩增,还具备极强的功能表达活性;基因拷贝数增加与亚功能分化,助力睡莲目植物适应水体环境光照散射、入射角度小、光照强度弱的特点,有效提升光能捕获与利用效率。
气孔退化与免疫负荷降低助力水生适应
为探究睡莲目植物气孔的演化历程,本研究选取 2 种沉水植物(合瓣莲、水盾草)与 2 种浮叶植物(红花睡莲、莼菜),解析并重构气孔发育相关遗传通路(图 4A、4B)。 比较基因组分析发现,四种睡莲目植物体内调控气孔形成的核心调控模块整体高度保守,主要包含类受体激酶 ERECTA 及其辅助因子 TMM、下游 YODA-MKK-MPK 信号级联通路、转录因子 SPCH 与 SCRM,以及终端调控因子 FAMA、FLP/CDKB1;1。 这说明睡莲目植物气孔发育的核心遗传框架在演化中十分稳定。 但所有受试物种均缺失多种气孔发育关键调控基因:例如启动气孔前体细胞分化的EPF2 、调控气孔分布间距的EPF1/2 ,以及调控细胞极性与不均等分裂的BASL 和POLAR (图 4D)。 关键基因的批量丢失,证明睡莲目植物的气孔调控网络在长期水生演化过程中发生了结构重塑。
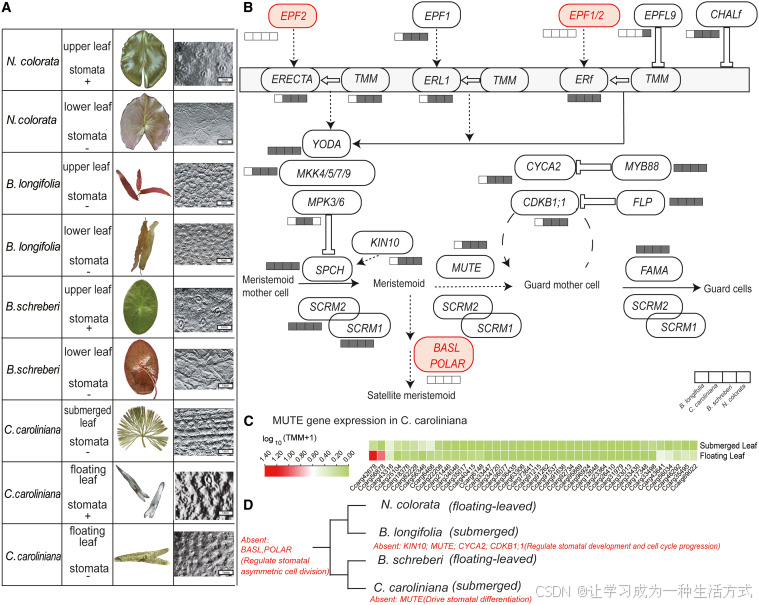
图 4 睡莲水生适应过程中气孔渐进式退化
(A) 四种代表性睡莲科植物(两种沉水植物、两种浮叶植物)的叶片类型与气孔分布情况。**"-"代表无气孔, "+"** 代表存在气孔。 (B) 气孔从分生组织母细胞逐步分化为保卫母细胞,最终形成保卫细胞的发育流程。方框从左至右依次对应合瓣莲、水盾草、莼菜、红花睡莲;灰色填充方框表示该物种含有对应基因。 (C) 水盾草叶片中MUTE 基因的表达水平。 (D) 图中红色标注基因为物种发生丢失的基因。
不同物种的气孔表型存在明显生态分化:水盾草与合瓣莲的沉水叶完全无气孔,而红花睡莲、莼菜的浮叶上表皮则保留气孔结构。转录组分析进一步揭示了该分化背后的分子机制:以水盾草为例,其基因组中气孔分化关键基因MUTE 序列完整,但在沉水叶中几乎不表达,仅在浮叶中呈现微量表达(图 4C、附图 1)。 这表明沉水叶气孔缺失并非由核心基因丢失造成,而是基因转录表达受到抑制所致。
其中水盾草的演化特征尤为特殊:自然环境下该物种极少形成浮叶,仅开花期会长出少量气孔稀疏的浮叶。该物种虽经历多次全基因组加倍事件,但沉水叶的气孔发育通路整体处于沉默状态,体现出基因序列保留与功能表达相互脱耦 的演化特征。 综上,睡莲目植物沉水叶气孔退化并非基因缺失导致,而是通过转录调控重编程 实现。 这种功能重塑模式代表了一种独特的水生适应演化策略:物种保留气孔发育的全套遗传基础,再依据器官类型与生存环境灵活调控基因表达,在维持基因组稳定性的同时,实现植株表型的可塑性调控。
本研究进一步系统统计对比了各类代表性维管植物体内核苷酸结合域富亮氨酸重复基因(NLR,抗病基因)的拷贝数量。 整体来看,裸子植物与绝大多数被子植物均含有数百至上千个 NLR 基因;多数单子叶、真双子叶植物的 NLR 基因拷贝数高于被子植物整体中位值,不同类群间数量差异显著。 与之形成鲜明对比的是,所有睡莲目受试物种的 NLR 基因数量均维持在极低水平。 即便身为睡莲目中基因组庞大、多倍化程度较高的水盾草,其 NLR 基因数量也远低于被子植物中位值(红色虚线),数量甚至接近或低于部分寄生植物、特化生态型植物。 上述结果证实,相较于绝大多数被子植物类群,睡莲目植物发生了显著的NLR 抗病基因家族收缩 (附表 6、附表 7)。
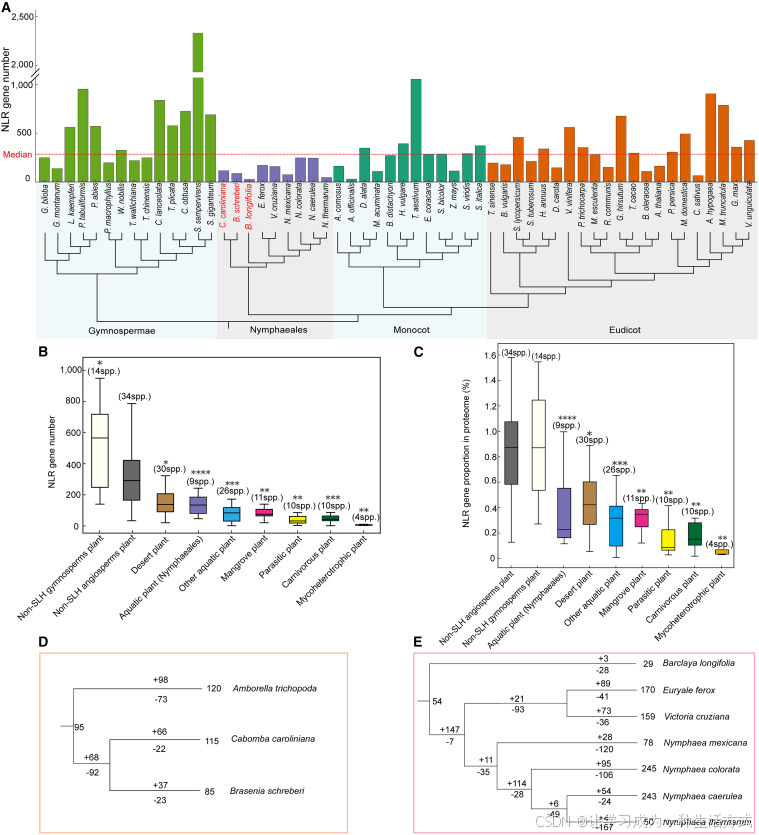
图 5 睡莲目植物抗病 NLR 基因家族收缩
(A) 不同植物类群 NLR 基因数量对比。本研究共鉴定 56 种代表性植物的 NLR 基因家族,包含 14 种裸子植物、9 种睡莲目植物(2 种沉水植物、7 种浮叶植物)、12 种单子叶植物及 21 种真双子叶植物。水盾草基因组体量偏大,但 NLR 基因数量仍低于整体中位值,证实睡莲目植物普遍存在 NLR 基因收缩现象(附图 3--- 附图 5)。
(B) 不同生态类型植物的 NLR 基因数量分布。分组包含:睡莲目水生植物 9 种、其他水生植物 26 种、寄生植物 10 种、食虫植物 10 种、菌异养植物 4 种、荒漠植物 30 种、红树林植物 11 种、非特化生境裸子植物 14 种、非特化生境被子植物 34 种。 特化生境植物 指适应特殊生存环境的物种,涵盖水生、寄生、食虫、菌异养、荒漠、红树林植物;非特化生境被子植物即无上述特殊适应特征的被子植物。 箱线图中线代表中位数,箱体边界为四分位距,须线代表 1.5 倍四分位距(离群值未展示)。采用双侧曼 - 惠特尼 U 检验分析非特化被子植物与其余类群的差异,多重检验采用本雅明尼 - 霍赫伯格错误发现率法校正P 值(** 校正后P <0.01;*校正后P <0.001;**校正后P <0.0001)。
(C) 各类植物基因组中 NLR 基因占全部蛋白编码基因的比例分布,物种分组与统计方法同 (B)。
(D) 睡莲科与无油樟 NLR 基因演化动态,分支上 **+代表基因复制事件, -** 代表基因丢失事件。
(E) 睡莲科植物 NLR 基因家族演化历程。
为进一步探究 NLR 基因演化与生态环境的关联,本研究依据生存习性对植物进行生态类群划分。统计结果表明,水生植物整体 NLR 基因数量显著低于陆生植物,其中睡莲目植物在所有水生类群中基因拷贝数处于极低水平。 从 NLR 基因占蛋白编码基因总数的占比来看,水生植物整体占比远低于陆生植物,睡莲目在所有受试植物类群中排名靠后。 以上结果说明水生环境对植物 NLR 抗病基因形成强烈的负向选择压力,睡莲目植物该基因家族收缩趋势最为显著。
基于系统发育的基因家族演化分析也印证了这一结论:睡莲目共同祖先分支处已发生大规模 NLR 基因缩减,莼菜、水盾草等多个物种均延续这一演化特征。 睡莲科内部不同物种演化模式存在分化,既有基因家族收缩,也存在物种特异性基因扩张(如合瓣莲、芡实)。整体而言,睡莲目 NLR 基因呈整体缩减趋势,类群内部演化差异明显。
结合水生适应的生态背景可推断:陆生植物主要依靠扩增 NLR 基因家族抵御多样病原菌,而睡莲目植物演化出了截然不同的防御策略。 物种体内 NLR 介导的免疫通路衰退,由受体激酶、GDSL 脂肪酶、胶质合成相关基因等家族扩张进行功能代偿,以此完成水生环境下的生物胁迫应答。
黄酮类物质代谢通路适应性调控助力水生逆境耐受
研究发现,儿茶素合成通路相关基因存在明显的组织特异性表达特征:该类基因在水盾草根系、莼菜分枝茎中协同上调表达。 上述组织多为地下组织或近水支撑组织,长期直面各类水生环境胁迫,说明儿茶素合成通路受空间特异性调控,并非在所有组织中持续恒定表达。
在转录水平上,处于黄酮代谢无色花青素分支关键节点的DFR、LAR、ANS、ANR 基因,集中在水生逆境相关组织中协同高表达(图 6)。 这类基因共同决定无色花青素流向黄烷 - 3 - 醇合成的代谢分配方向,其组织特异性激活表明,水生植物可通过精准调控代谢通路关键分支节点的物质流向,实现儿茶素合成通路的定向调控。 这种单一通路节点的协同调控模式,属于靶向性通路层级调控,而非整个黄酮代谢网络的泛化改变,体现出植物在保守黄酮合成骨架基础上进行适应性微调的演化特点。
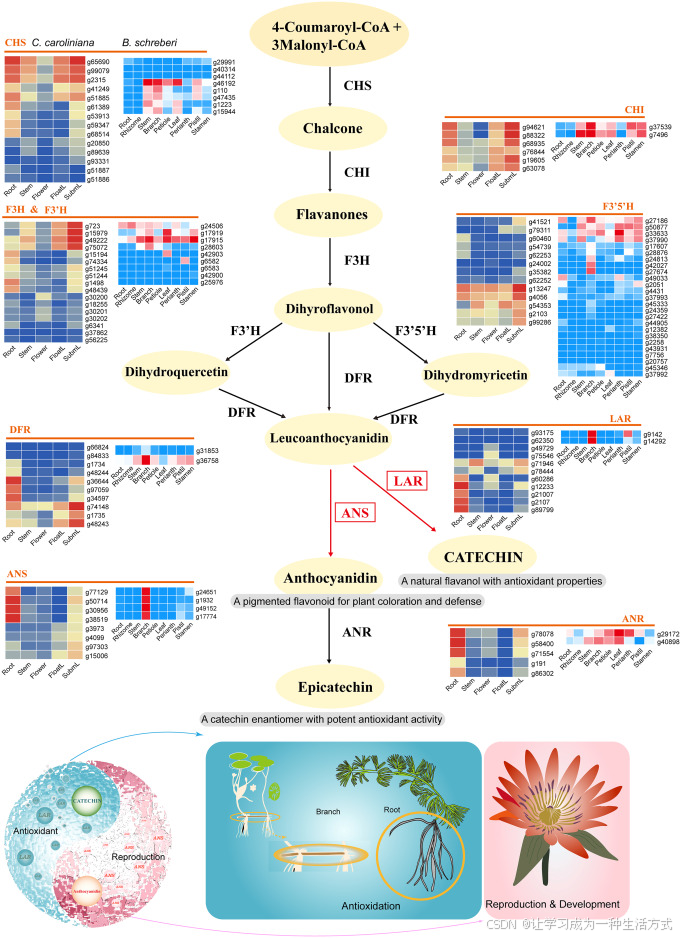
图 6 水盾草与莼菜的儿茶素合成通路及相关基因表达特征
左右两侧热图分别展示水盾草、莼菜体内儿茶素合成通路相关基因的表达模式。其中DFR、LAR、ANS、ANR 在水盾草根部特异性高表达,DFR、LAR、ANS 在莼菜分枝茎中高表达。下方示意图表明,儿茶素在水盾草根部与莼菜分枝茎中大量积累。花青素作为无色花色素向表儿茶素转化过程中的中间产物,还参与调控植物花器官的生殖生长与发育进程。
基因缩写释义 CHS:查尔酮合酶 CHI:查尔酮异构酶 F3H:黄烷酮 3 - 羟化酶 F3′H:黄酮类 3′- 羟化酶 F3′5′H:黄酮类 3′,5′- 羟化酶 DFR:二氢黄酮醇 4 - 还原酶 LAR:无色花青素还原酶 ANS:花青素合成酶 ANR:花青素还原酶
研究发现,DFR、LAR、ANS、ANR 基因上调表达的同时,普遍伴随大量内含子保留 可变剪接事件(附图 11)。该剪接形式在植物根、茎组织中稳定存在,在其他组织中则显著减少甚至消失。 这一关联特征说明,内含子保留可作为可变剪接的一种调控方式,能够调控儿茶素合成相关基因的转录本稳定性、翻译效率,或是改变编码酶蛋白的功能活性。 这类基因在特定组织中富集内含子保留现象,并非无意义的剪接异常,而是水生植物特有的转录调控层次,可精细调控水生组织内次生代谢通路的运转效率。
代谢通路重塑助力水生环境适应
目前学界尚未发现可作为植物水生适应标志性特征的小分子代谢物。为此,本研究对比分析两种沉水植物(水盾草、合瓣莲)与两种浮叶植物(莼菜、红花睡莲)的代谢组图谱。 气相色谱 - 质谱联用(GC-MS)检测结果显示,角鲨烯 在沉水植物体内含量显著更高,其叶片中角鲨烯积累量至少为浮叶植物的 4 倍(图 7A)。
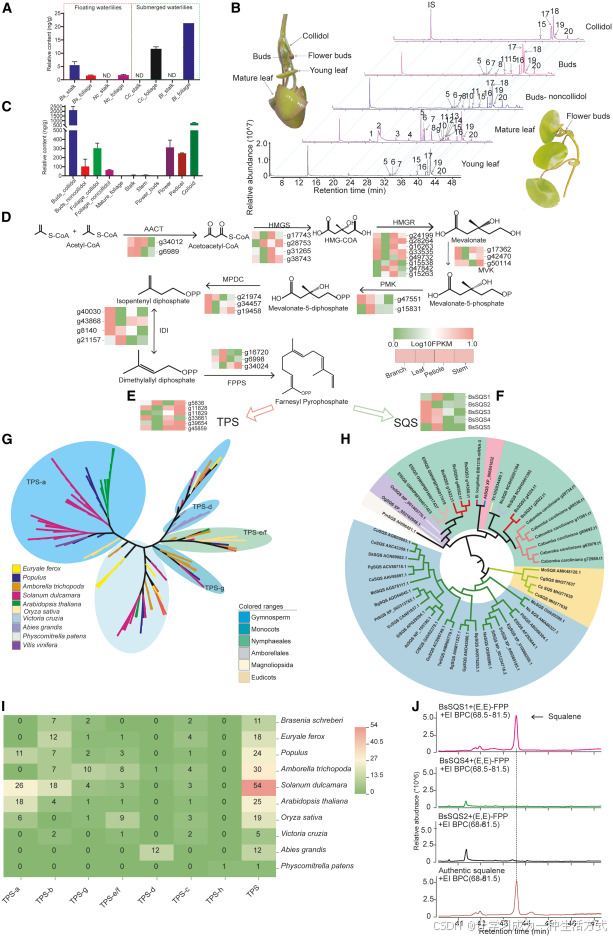
图 7 萜类 - 角鲨烯代谢通路重塑介导睡莲水生适应
(A) 莼菜、合瓣莲、水盾草、红花睡莲叶柄与叶片中的角鲨烯含量。 (B) 莼菜不同组织、花芽、黏液、成熟叶、幼叶的挥发性物质气相色谱图。 (C) 角鲨烯在莼菜不同组织中的相对富集含量。 (D) 甲羟戊酸合成通路相关编码酶基因的表达模式。 (E) 莼菜萜类合酶基因在分枝、叶片、叶柄、茎中的表达特征。 (F) 莼菜角鲨烯合酶基因在分枝、叶片、叶柄、茎中的表达特征。 (G) 莼菜萜类合酶与典型植物萜类合酶的系统发育树。 (H) 莼菜角鲨烯合酶基因系统发育树。 (I) 莼菜及其他植物萜类合酶在七大亚家族中的基因分布情况。 (J) 体外酶促反应产物鉴定:BsSQS1 (g2022)、BsSQS4 (g46562)、BsSQS2 (g4324) 以反式法尼基焦磷酸为底物的催化产物分析。
角鲨烯标准品购于商业试剂公司。 图 B 色谱峰对应物质:1 - 乙烯基愈创木酚;2 - 焦性没食子酸;3 - 菜籽素;4-3 - 氧代 -α- 紫罗兰醇;5 - 新植二烯;6-3,7,11,15 - 四甲基 - 2 - 十六碳烯 - 1 - 醇;7 - 十六烷酸;8 - 十七碳三十五烯;9 - 二十七烷;10 - 植醇;11 - 亚油酸乙酯;12 - 亚麻酸;13 - 硬脂酸;14 - 十六烷酰胺;15 - 二十三烷;16 - 角鲨烯;17 - 顺式 - 9 - 十八碳烯酰胺;18 - 十八烷酰胺;19 - 二十五烷。
缩写释义: HMG-CoA:3 - 羟基 - 3 - 甲基戊二酰辅酶 A DMAPP:二甲基丙烯焦磷酸 FPP:法尼基焦磷酸
本研究以莼菜为代表,对其幼嫩茎段、花、花芽、根状茎、成熟叶、叶柄、花梗七种组织开展挥发性代谢物检测。结果显示,脂肪酸衍生物与角鲨烯是其主要挥发性成分;角鲨烯在幼嫩组织与黏液中含量极高,在成熟叶片中仅微量存在(图 7B、7C)。
基因组层面发现,莼菜体内 ** 萜类合酶(TPS)** 基因发生大规模丢失,尤以负责合成单萜、倍半萜的 TPS-a 与 TPS-e/f 亚家族丢失最为显著(图 7I)。转录组数据证实,经甲羟戊酸(MVA)通路生成法尼基焦磷酸(FPP)后,代谢流不再流向萜类合成分支,而是在高表达角鲨烯合酶(SQS)基因的驱动下,大量转向角鲨烯合成途径(图 7D)。
研究筛选得到 3 个在多组织中高表达的莼菜角鲨烯合酶基因,系统发育分析显示其聚类于单子叶植物演化分支;体外酶活实验证实,BsSQS1 可催化底物反式法尼基焦磷酸生成角鲨烯(图 7J)。
综上,萜类合酶基因大量丢失重塑了植物体内代谢流向,促使角鲨烯在幼嫩组织与黏液中大量积累。该代谢转向可对植物幼嫩脆弱组织起到保护作用,是莼菜形成独特水生生态适应性的重要分子基础。由此可知,演化过程中萜类合酶基因的丢失,让水生植物将物质与能量优先用于合成角鲨烯,最终助力其适应水生生存环境。
讨论
多倍化、染色体重排与功能重塑协同驱动水生适应
全基因组加倍(WGD)是早期被子植物演化创新的核心驱动力,基因冗余可推动基因功能分化,助力植物适应新生境。本研究证实,睡莲目祖先物种约在 1.2 亿年前发生一次古老全基因组加倍事件(π 事件),为睡莲科与水盾草科积累了充足遗传冗余,为后续基因功能重塑奠定基础。 其中水盾草还经历二次二倍化、四倍化等多次基因组加倍事件,体现出沉水植物基因组持续动态重组的演化特征,使其更好适配水下生长环境。这表明多倍化仅提供遗传进化潜能,物种适应性表型形成与类群分化,最终依赖加倍基因的保留、丢失与功能分化。
染色质结构变异在睡莲目演化进程中发挥关键作用,是植物基因组响应环境选择压力的重要方式。系统发育基因组学与共线性分析表明,睡莲目祖先物种经历大量染色体断裂融合、近着丝粒融合、反向转座等结构重排事件。 这类结构变异可调控基因表达与代谢通路,助力植物适应水生环境。水盾草及水盾草科植物复杂的基因组结构,可优化体内物质能量分配,降低高耗能免疫通路的资源消耗。 通常极端环境易促进植物多倍化发生,而水生环境整体环境相对稳定,但水盾草科植物仍发生多次全基因组加倍,说明水生植物基因组演化规律有别于传统认知的稳态演化模型。 多次基因组加倍并未带来植株形态结构复杂化与物种多样性提升,如水盾草花器官结构简单,与睡莲科大而复杂的花器官形成鲜明对比。染色体重排主要驱动光合作用、物质代谢相关功能模块发生表达调控改变,最终塑造出水生植物特有的沉水生长性状。
功能重塑是植物实现水生适应的核心途径,基因加倍(含多倍化)是其重要助力。水生植物面临光照条件改变、气体交换受阻、养分获取受限等特殊生存胁迫,亟需快速完成基因组层面适应性改造。 基因家族扩张分析显示,睡莲目植物光合作用、叶片形态建成、水分胁迫应答相关基因家族(如捕光复合物基因)显著扩增。这类基因扩张大多源于多倍化后基因保留,同时也无法排除串联重复、片段重复等其他复制方式的作用。 基因家族的扩张与收缩,重塑了水生植物体内代谢与调控网络,使其能在复杂水下环境中高效捕获光能、正常生长发育。与此同时,NLR 等免疫相关基因家族收缩,大幅降低植株免疫代谢消耗,节省更多能量用于生长发育与其他适应性生理过程。 莼菜体内萜类合酶基因大量丢失,使代谢流定向流向角鲨烯合成通路,角鲨烯在幼嫩组织与黏液中大量富集,为幼嫩组织提供防护作用。以上实例均证实,基因功能重塑是植物实现水生适应性演化的重要核心机制。
以水盾草体内捕光复合物基因与抗病 NLR 基因为例,解析染色体结构变异与全基因组加倍对基因家族演化的影响:水盾草全部 131 个捕光复合物基因均位于基因组共线性区段内,而合瓣莲仅有半数左右该基因分布于共线性区域,说明捕光复合物基因家族扩张与古老全基因组加倍、大规模染色体片段复制密切相关。 这类基因散布于多条染色体,大量分布在染色体断裂与重排区域,证明基因组结构变异为基因保留与拷贝数扩增提供了遗传基础。 依据共线性区段同义替换率(Ks)溯源水盾草 131 个捕光复合物基因演化起源:9 个起源于最古老 π 事件全基因组加倍,49 个起源于后续 κ 事件加倍,70 个起源于晚期加倍事件,全部基因均参与近期四倍化事件。 该类基因分布范围广,受染色体重排影响深刻,证明基因组结构变异重塑其分布模式,推动基因拷贝数增加,提升植株光合利用效率。 反观 NLR 抗病基因:π 事件加倍后保留 25 个,κ 事件仅 3 个基因发生复制扩张,可见全基因组加倍对不同基因家族的演化影响存在显著差异,对捕光基因扩增作用显著,对抗病基因无明显扩增效应。 综上,不同基因家族在连续多倍化事件中呈现截然不同的保留与扩张模式,证实基因组结构变异可差异化调控各类基因家族演化进程,也阐明了全基因组加倍与染色体重排通过驱动捕光基因扩张,赋予植物更强水生环境光合适应能力。
水生植物能量获取与免疫防御演化策略
水生植物不仅受光照、气体交换条件限制,还需应对各类环境胁迫与病原菌侵染。睡莲目植物捕光复合物基因显著扩张,是沉水植物适应弱光环境、提升光能捕获能力的关键。 水盾草依靠大量扩增的捕光基因,可在弱光水体环境中高效捕获光能,保障沉水状态下光合作用正常进行。 与此同时,NLR 免疫基因家族收缩是植物适应性降低免疫消耗的直接体现,植株将更多物质能量分配至生长发育等核心生命活动。 水生环境病原菌种类、侵染密度与陆生环境差异较大,植物维持大规模 NLR 介导免疫通路的选择压力大幅减弱。水生植物下调免疫资源投入,将能量优先用于生长与繁殖,大幅提升其在水生生态位中的环境适合度。
气孔发育重塑与表达调控重编程
气孔发育通路解析结果证实,沉水植物与浮叶植物气孔发育调控模式存在巨大分化。水盾草、莼菜等全沉水植物叶片完全无气孔结构,红花睡莲、莼菜浮叶上表皮则正常形成气孔。 沉水植物基因组完整保留全套气孔发育调控基因,说明沉水叶气孔表型缺失并非基因丢失所致,而是基因转录表达受到抑制。 以MUTE 基因为代表的气孔发育关键基因在沉水叶中表达沉默,直接阻断气孔细胞分化进程。 这种 "基因序列完整保留、表达转录受抑" 的调控模式,让水生植物在保留表型可塑性遗传潜能的前提下,通过调控重编程实现器官性状退化。 水盾草经历多次全基因组加倍,拥有大量气孔发育相关冗余基因,但这类复制基因大多始终处于表达沉默状态。 这说明多倍化可稳定维持体内调控网络框架,无需丢失功能基因即可完成生理通路改造。水盾草仅在特定时期长出少量带气孔浮叶,实现气孔功能条件性启动,体现出水生植物在维持基因组稳定与灵活调整表型之间形成的精密平衡策略。
代谢流向重塑与水生适应机制
本研究整合多组学分析证实,儿茶素合成通路是基部睡莲类植物演化中一项尚未被充分重视的水生适应生化革新。与分布广泛的普通黄酮类物质不同,儿茶素具有极强的组织特异性,主要富集于地下器官或近水组织中,如水盾草根系、莼菜茎秆。 合成通路相关基因协同上调表达,加之产物的组织定向积累,表明儿茶素合成是植物主动调控形成的器官特异性适应性状,并非黄酮类基础代谢产生的副产物。
从演化层面来看,合成儿茶素与各类黄酮物质的苯丙烷通路,是植物演化早期就已形成并高度保守的古老代谢通路。轮藻及早期陆生植物的酚类物质代谢,长期受紫外线辐射、干旱胁迫、氧化应激等环境压力塑造,逐步形成功能多元、来源广泛的代谢体系。 据此推断,睡莲目中的儿茶素合成通路并非该类群独有的全新演化产物,而是祖先植物原有保守代谢通路发生流向重编程与组织表达重分配 后形成的适应性结果。
分子机制层面,基部睡莲植物体内儿茶素大量积累,核心在于黄酮代谢通路无色花青素分支节点处的代谢流重定向。 LAR、ANS、ANR 基因协同激活,推动碳源物质分别流向反式与顺式黄烷 - 3 - 醇合成支路,同步生成儿茶素与表儿茶素两类活性物质。 这类基因还普遍存在内含子保留可变剪接事件,构成转录后调控层级,助力植物在复杂多变的水生环境中精准调控儿茶素合成速率。 功能上,组织特异性积累的儿茶素与表儿茶素可增强沉水组织对低氧诱导氧化胁迫的耐受能力,稳固细胞结构,同时抑制微生物侵染;生长在水体近表层的器官则依靠多酚类物质提升机械韧性,抵御水流冲击与水体生物附着危害。
综上研究结果证实,睡莲植物借助代谢分支节点调控与可变剪接调控,改造利用原有保守黄酮代谢通路,从而满足水生环境下的生理生存需求。 这种多层级调控模式,体现出保守代谢通路如何在全新环境压力下完成定向功能优化,也从基因组与生化层面阐明了睡莲目植物水生适应的分子基础。 组织特异性基因表达与内含子保留介导的转录后调控,共同实现次生代谢通路的适应性微调,大幅提升植物水生逆境耐受能力。
植物适应淹水与沉水环境还具备多重形态与生化适应策略,包括通气组织形成、不定根发生、无氧代谢途径激活,以及激素与活性氧信号通路调控等。 活性氧既是通气组织发育过程中的信号分子,同时也会造成生理胁迫;甾醇与细胞膜脂质能够调控细胞膜流动性、蛋白功能及激素受体定位,因此代谢流向偏向角鲨烯与甾醇合成,可直接影响植物对淹水低氧环境的应答能力。 莼菜体内萜类合酶基因大量丢失,使得萜类代谢流全面转向角鲨烯合成,角鲨烯在幼嫩组织与黏液中大量富集并发挥防护作用;转录组数据证实甲羟戊酸通路与角鲨烯合酶基因持续高表达,确立了角鲨烯合成在其水生适应中的核心地位。 该案例充分说明,代谢流向重塑能够弥补基因丢失带来的功能缺失,是水生植物特有的生化适应演化策略。
睡莲目水生适应演化启示
本研究表明,睡莲目植物各类水生适应性性状,均是植物向全沉水、半沉水生境转变过程中逐步形成的次级适应特征 。 这类基部被子植物的水生适应性演化,主要依靠基因丢失、基因家族扩张与表达调控重塑实现,并非单纯因进入水生环境而从头诞生全新基因组功能。 尽管睡莲目物种普遍经历基因组加倍与染色体重排,但这类基因组变异主要用于调整体内物质资源分配模式、优化光合效率、完善逆境胁迫应答体系,并非仅由水生环境单一胁迫主导形成。 这也证实,睡莲目典型水生性状的形成,本质是物种长期适应水生环境所产生的一系列适应性演化改变。
整体研究证实,多倍化为睡莲目植物后续水生适应相关的基因功能重塑提供了充足基因组遗传基础。 整合睡莲目物种基因组、转录组与代谢组数据可知,植物水生适应是遗传冗余、基因家族演化、代谢通路重编程与免疫体系优化多方协同作用的结果。 物种通过改变基因拷贝数与调控基因转录表达,精准调控光能捕获效率、物质代谢流向与免疫防御策略,最终顺利在沉水环境中正常生长繁衍。 本研究不仅阐明了水生适应的内在分子机制,也为解析睡莲目植物生态与演化历程提供新思路,明确了多倍化与表达调控可塑性在塑造水生生活习性中的重要演化潜力。
研究局限性
本研究所用基因组组装序列均已达到染色体水平,但仍存在一定不足: 第一,虽然核心假染色体组装质量良好,但受二代短读长测序与 Hi-C 技术本身限制,着丝粒、端粒等高度重复区域仍未实现完整组装; 第二,本次基因模型注释主要依托同源比对与转录组数据辅助完成,部分物种特有基因、快速演化基因仍存在注释不完整的问题; 第三,本研究比较基因组分析仅依托现有已发表被子植物基因组数据,后续纳入更多水生被子植物基因组资源,能够进一步完善水生适应相关演化结论; 第四,本研究筛选得到大量水生适应候选基因,其具体生物学功能仍有待后续实验开展体内外功能验证。
STAR★Methods
Key resources table
|------------------------------------------------------------|----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Deposited data |||
| Cabomba caroliniana genome sequencing data | This paper | NGDC/GSA: PRJCA030329 |
| Cabomba caroliniana transcriptome sequencing data | This paper | NGDC/GSA: PRJCA031632 |
| Cabomba caroliniana light-treatment transcriptome data | This paper | NGDC/GSA: PRJCA042289 |
| Barclaya longifolia genome sequencing data | This paper | NGDC/GSA: PRJCA030483 |
| Barclaya longifolia transcriptome sequencing data | This paper | NGDC/GSA: PRJCA031634 |
| Brasenia schreberi genome sequencing data | This paper | NGDC/GSA: PRJCA030328 |
| Brasenia schreberi transcriptome sequencing data | This paper | NGDC/GSA: PRJCA030291 |
| Software and algorithms |||
| NextDenovo v2.5 | Hu et al. href="#bib28" 28 | GitHub - Nextomics/NextDenovo: Fast and accurate de novo assembler for long reads · GitHub |
| NextPolish v1.4 | Hu et al. href="#bib29" 29 | GitHub - Nextomics/NextPolish: Fast and accurately polish the genome generated by long reads. · GitHub |
| Hifiasm v0.19.5 | Cheng et al. href="#bib30" 30 | Releases · chhylp123/hifiasm · GitHub |
| Haphic v1.0.6 | Zeng et al. href="#bib31" 31 | GitHub - zengxiaofei/HapHiC: HapHiC: a fast, reference-independent, allele-aware scaffolding tool based on Hi-C data · GitHub |
| Juicer | Robinson et al. href="#bib32" 32 | GitHub - aidenlab/juicer: A One-Click System for Analyzing Loop-Resolution Hi-C Experiments · GitHub |
| Juicerbox | Durand et al. href="#bib33" 33 | GitHub - aidenlab/Juicebox: Visualization and analysis software for Hi-C data - · GitHub |
| ALLHiC v0.9.8 | Zhang et al. href="#bib34" 34 | ALLHiC/allhic.v0.9.8 at master · tangerzhang/ALLHiC · GitHub |
| RepeatModeler v2.0.4 | Flynn et al. href="#bib35" 35 | Releases · Dfam-consortium/RepeatModeler · GitHub |
| RepeatMasker v4.1.5 | Chen et al. href="#bib36" 36 | RepeatMasker Home Page |
| BRAKER v3.0.3 | Hoff et al. href="#bib37" 37 | Releases · Gaius-Augustus/BRAKER · GitHub |
| GeneMark-EX | Bruna et al. href="#bib38" 38 | GeneMark gene prediction |
| Augustus | Stanke et al. href="#bib39" 39 | Augustus: gene prediction |
| Tandem Repeats Finder v4.09 | Benson et al. href="#bib40" 40 | Tandem Repeats Finder |
| LTR_Finder | Xu et al. href="#bib41" 41 | GitHub - oushujun/LTR_FINDER_parallel: A parallel wrapper for LTR_FINDER · GitHub |
| BUSCO v5.4.7 | Simao et al. href="#bib42" 42 | BUSCO - from QC to gene prediction and phylogenomics |
| Merqury v1.4.1 | Rhie et al. href="#bib43" 43 | https://github.com/marbl/merqury |
| OrthoFinder v2.5.2 | Emms et al. href="#bib44" 44 | https://github.com/davidemms/OrthoFinder/releases |
| PAML v4.9 | Yang et al. href="#bib45" 45 | https://github.com/abacus-gene/paml |
| iTOL | Letunic et al. href="#bib46" 46 | https://itol.embl.de/ |
| WGDI | Sun et al. href="#bib47" 47 | GitHub - SunPengChuan/wgdi: WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes · GitHub |
| HMMER v3.4 | Finn et al. href="#bib48" 48 | HMMER |
| OATK v1.0 | Zhou et al. href="#bib49" 49 | https://github.com/c-zhou/oatk |
| GetOrganelle v1.7.7 | Jin et al. href="#bib50" 50 | https://github.com/Kinggerm/GetOrganelle |
| minimap2 v2.28 | Li et al. href="#bib51" 51 | https://github.com/lh3/minimap2 |
| miniasm v0.3 | Li et al. href="#bib52" 52 | https://github.com/lh3/miniasm |
| Racon v1.4.3 | Vaser et al. href="#bib53" 53 | Releases · isovic/racon · GitHub |
| CHLOROBOX | Tillich et al. href="#bib54" 54 | https://chlorobox.mpimp-golm.mpg.de/ |
| ARAGORN v1.2.38 | Laslett et al. href="#bib55" 55 | https://www.trna.se/ARAGORN/Downloads/ |
| ARWEN v1.2.3 | Laslett et al.[55](#bib28" 28 GitHub - Nextomics/NextDenovo: Fast and accurate de novo assembler for long reads · GitHub NextPolish v1.4 Hu et al. href="#bib29" 29 GitHub - Nextomics/NextPolish: Fast and accurately polish the genome generated by long reads. · GitHub Hifiasm v0.19.5 Cheng et al. href="#bib30" 30 Releases · chhylp123/hifiasm · GitHub Haphic v1.0.6 Zeng et al. href="#bib31" 31 GitHub - zengxiaofei/HapHiC: HapHiC: a fast, reference-independent, allele-aware scaffolding tool based on Hi-C data · GitHub Juicer Robinson et al. href="#bib32" 32 GitHub - aidenlab/juicer: A One-Click System for Analyzing Loop-Resolution Hi-C Experiments · GitHub Juicerbox Durand et al. href="#bib33" 33 GitHub - aidenlab/Juicebox: Visualization and analysis software for Hi-C data - · GitHub ALLHiC v0.9.8 Zhang et al. href="#bib34" 34 ALLHiC/allhic.v0.9.8 at master · tangerzhang/ALLHiC · GitHub RepeatModeler v2.0.4 Flynn et al. href="#bib35" 35 Releases · Dfam-consortium/RepeatModeler · GitHub RepeatMasker v4.1.5 Chen et al. href="#bib36" 36 RepeatMasker Home Page BRAKER v3.0.3 Hoff et al. href="#bib37" 37 Releases · Gaius-Augustus/BRAKER · GitHub GeneMark-EX Bruna et al. href="#bib38" 38 GeneMark gene prediction Augustus Stanke et al. href="#bib39" 39 Augustus: gene prediction Tandem Repeats Finder v4.09 Benson et al. href="#bib40" 40 Tandem Repeats Finder LTR_Finder Xu et al. href="#bib41" 41 GitHub - oushujun/LTR_FINDER_parallel: A parallel wrapper for LTR_FINDER · GitHub BUSCO v5.4.7 Simao et al. href="#bib42" 42 BUSCO - from QC to gene prediction and phylogenomics Merqury v1.4.1 Rhie et al. href="#bib43" 43 https://github.com/marbl/merqury OrthoFinder v2.5.2 Emms et al. href="#bib44" 44 https://github.com/davidemms/OrthoFinder/releases PAML v4.9 Yang et al. href="#bib45" 45 https://github.com/abacus-gene/paml iTOL Letunic et al. href="#bib46" 46 https://itol.embl.de/ WGDI Sun et al. href="#bib47" 47 GitHub - SunPengChuan/wgdi: WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes · GitHub HMMER v3.4 Finn et al. href="#bib48" 48 HMMER OATK v1.0 Zhou et al. href="#bib49" 49 https://github.com/c-zhou/oatk GetOrganelle v1.7.7 Jin et al. href="#bib50" 50 https://github.com/Kinggerm/GetOrganelle minimap2 v2.28 Li et al. href="#bib51" 51 https://github.com/lh3/minimap2 miniasm v0.3 Li et al. href="#bib52" 52 https://github.com/lh3/miniasm Racon v1.4.3 Vaser et al. href="#bib53" 53 Releases · isovic/racon · GitHub CHLOROBOX Tillich et al. href="#bib54" 54 https://chlorobox.mpimp-golm.mpg.de/ ARAGORN v1.2.38 Laslett et al. href="#bib55" 55 https://www.trna.se/ARAGORN/Downloads/ ARWEN v1.2.3 Laslett et al.55 https://mybiosoftware.com/arwen-1-2-3-trna-detection-in-metazoan-mitochondrial-sequences.html EDTA v2.2.2 Ou et al. href="#bib56" 56 https://github.com/oushujun/EDTA/releases Other Custom scripts for polyploidization and karyotype analysis This paper GitHub - SunPengChuan/Angiosperm-karyotype-evolution: Angiosperm karyotype evolution · GitHub DNeasy Plant Maxi Kit QIAGEN Cat#69163 TRIzol reagent Thermo Fisher Cat#15596026 Iso-Seq Express Oligo Kit PacBio Cat#101-683-100 SMRTbell Express Template Prep Kit 2.0 PacBio Cat#100-938-900 pET32a vector Merck Cat#69015-3 E. coli BL21 (DE3) Thermo Fisher Cat#EC0114) | https://mybiosoftware.com/arwen-1-2-3-trna-detection-in-metazoan-mitochondrial-sequences.html |
| EDTA v2.2.2 | Ou et al. href="#bib56" 56 | https://github.com/oushujun/EDTA/releases |
| Other |||
| Custom scripts for polyploidization and karyotype analysis | This paper | GitHub - SunPengChuan/Angiosperm-karyotype-evolution: Angiosperm karyotype evolution · GitHub |
| DNeasy Plant Maxi Kit | QIAGEN | Cat#69163 |
| TRIzol reagent | Thermo Fisher | Cat#15596026 |
| Iso-Seq Express Oligo Kit | PacBio | Cat#101-683-100 |
| SMRTbell Express Template Prep Kit 2.0 | PacBio | Cat#100-938-900 |
| pET32a vector | Merck | Cat#69015-3 |
| E. coli BL21 (DE3) | Thermo Fisher | Cat#EC0114 |
实验模型与研究材料
植物材料
莼菜与水盾草采集自中国天然野生生境,长叶合瓣莲采集自印度尼西亚雅加达。实验统一选用生长周期 6 个月的植株开展基因组 DNA 与总 RNA 提取;依据实验需求分别采集幼叶、根系、茎秆、叶柄及花器官等组织样品。 本研究采集物种均为非濒危野生植物,无需专项采集许可,植物凭证标本保存于海南大学。本研究不涉及人体受试者、实验动物及细胞系相关实验。
实验方法详情
基因组与转录组测序
分别取莼菜、水盾草幼嫩叶片提取基因组总 DNA。莼菜基因组测序采用牛津纳米孔长读长测序平台(ONT) ,构建高分子量 DNA 文库,于 PromethION/GridION ×5 平台完成长片段测序;同时依托 Illumina NovaSeq 平台构建插入片段约 350 bp 文库,产出 150 bp 双端短读长数据,用于提升基因组组装完整度与精准度。 水盾草采用PacBio HiFi 高保真长读长测序 。两种植物均选取幼叶依照标准流程构建 Hi-C 染色体构象捕获文库,借助 Illumina NovaSeq 平台测序(插入片段 300~600 bp),辅助完成染色体水平基因组挂载组装。
转录组测序取材涵盖根、茎、叶、花等多种组织,提取总 RNA 后通过琼脂糖凝胶电泳与微量分光光度计联合检测 RNA 完整性与纯度。合格样品构建 cDNA 文库,于 NovaSeq 平台测序,获得高质量转录组数据,用于基因预测、结构注释与基因表达量分析。
长叶合瓣莲取材自印尼雅加达 6 月龄植株,利用 QIAGEN 植物大型 DNA 提取试剂盒提取幼叶基因组 DNA,构建 ONT 长读长文库并完成上机测序;取 1.5 μg 基因组 DNA 片段化至 350 bp 左右构建 Illumina 文库,NovaSeq 平台产出 150 bp 双端数据,同步完成 Hi-C 文库构建与上机测序。 转录组层面采用 TRIzol 试剂或 CTAB - 氯化锂法提取叶片、根系、叶柄组织 RNA,严格筛选合格样本(OD260/280=1.8~2.2、OD260/230≥2、RNA 完整度指数 RIN≥8、总 RNA 量≥1 μg)进行文库构建。取 300 ng 合格 RNA 反转录合成 cDNA,借助 PacBio 全长转录组试剂盒建库,在 PacBio Sequel II 平台获取全长转录本序列;同时利用 oligo (dT) 磁珠富集 mRNA,随机引物反转录后于华大 DNBSEQ 平台测序产出短片段数据,补充完善基因注释与表达定量分析。
基因组组装与功能注释
莼菜依托 NextDenovo v2.5.2 组装 113 Gb ONT 长读长数据,获得高连续性基因组序列,再使用 NextPolish v1.4.1 结合 Illumina 短读长数据多轮纠错修正碱基错误;水盾草利用 Hifiasm v0.19.5 组装 151 Gb PacBio HiFi 精准读长序列;长叶合瓣莲组装流程一致,初组装完成后经五轮序列纠错,再通过 ALLHiC 软件结合 Hi-C 数据完成染色体层级组装,最终借助 Juicer 及 Juicerbox 软件人工校正基因组挂载结果。
重复序列注释:使用 RepeatModeler v2.0.4 构建物种特异性重复序列数据库,搭配 RepeatMasker v4.1.5 完成散在重复序列与低复杂度序列屏蔽。 基因结构注释:莼菜与水盾草采用 BRAKER v3.0.3 整合转录组数据、同源蛋白序列联合预测基因;长叶合瓣莲先通过 EviAnn 软件整合近缘物种同源序列与转录组数据构建高可信度基因集,再融合多款预测软件结果补充完善注释信息。 利用串联重复序列查找软件鉴定串联重复序列,LTR_FINDER 完成长末端重复反转座子注释;采用 BUSCO v5.4.7 选用陆生植物数据库评估基因组组装完整度,Merqury 软件评估基因组碱基准确度,为后续系统发育与功能基因组分析奠定数据基础。
系统发育树构建与分化时间估算
整合 34 种代表性植物蛋白序列,涵盖苔藓植物、裸子植物、基部被子植物(无油樟目、睡莲目)、单子叶植物、木兰类植物及真双子叶植物,全面覆盖被子植物各大演化类群。 通过 OrthoFinder v2.5.2 筛选单拷贝同源基因,完成多序列比对并剔除低质量序列,构建最大似然进化树;依托 TimeTree 化石校正数据库设置 10 个可靠化石校正点,运用 PAML 软件中 MCMCTree 贝叶斯模型估算物种分化时间,最终在 iTOL 在线平台美化进化树,明确睡莲目及基部被子植物演化时间脉络。
多倍化事件鉴定与祖先核型重构
利用自研脚本筛选最长转录本与染色体定位基因,借助 WGDI 软件完成种内与种间共线性区块比对,计算同源基因同义替换率 Ks 值并绘制 Ks 散点图;采用 YN00 算法校正不同物种同义替换速率差异,统一标准化 Ks 数值,精准区分物种分化事件与全基因组加倍事件。 结合已明确的多倍化事件与可靠进化拓扑结构,以近缘物种为外类群,依据最大简约法则重构睡莲目祖先染色体核型,系统鉴定染色体末端融合、嵌套染色体融合、染色体臂相互易位、染色体丢失与断裂等结构变异事件,相关详细分析流程已上传开源教程。
比较基因组学分析
选取 24 种代表性植物开展比较基因组研究,囊括无油樟、多种睡莲目植物、单子叶植物、真双子叶植物、木兰类植物及木兰藤目植物。 通过同源基因聚类筛选单拷贝同源基因,构建进化树并估算分化时间;利用 CAFE 软件分析基因家族扩张与收缩动态,筛选类群特异性功能变异基因;依托 Pfam 蛋白结构域数据库结合 HMMER 软件注释保守结构域,统计物种特有与共有功能结构域,挖掘睡莲目水生适应性演化相关候选基因与功能模块。
叶绿体与线粒体基因组组装注释
分别完成三种睡莲目植物质体基因组与线粒体基因组组装注释。水盾草叶绿体基因组利用 PacBio HiFi 数据结合专用组装软件完成组装,莼菜叶绿体基因组依托二代短读长数据使用 GetOrganelle 软件组装,借助 Bandage 软件验证环状基因组完整性。 线粒体基因组先通过长读长序列完成初步组装,经多轮纠错与二代数据碱基抛光提升序列精准度。所有细胞器基因组统一在 CHLOROBOX 平台完成基因功能注释,借助多款软件精准预测转运 RNA 基因,最终导出标准基因注释文件并绘制细胞器基因组物理图谱,叶绿体图谱优化后可直接用于论文配图。
重复序列精细注释
使用串联重复序列查找软件设定标准化参数鉴定简单串联重复序列;采用同源预测 + 从头预测 双策略注释转座元件:一方面依托 RepeatMasker 比对 Repbase 已知重复序列数据库,注释已知转座子;另一方面利用 EDTA 整合分析流程构建物种专属非冗余转座子数据库,精准区分自主型与非自主型 Helitron 转座子。 完成序列注释后统一筛选相似度阈值,整合所有转座元件统计序列总长与丰度;借助脚本解析重复序列比对结果,依据序列分化程度结合物种演化速率公式,估算各类转座子远古插入分化时间。
莼菜挥发性物质分析与角鲨烯合酶体外酶活验证
春季采集新鲜莼菜幼嫩组织液氮速冻低温保存;液氮研磨后加入含内标正辛醇的乙酸乙酯溶液常温静态浸提,采用气相色谱 - 串联质谱联用技术测定挥发性组分,色谱柱选用 HP-5MS,设定标准升温程序与质谱检测参数。 结合 NIST14 标准质谱图库完成物质定性,内标法实现物质定量;以变量重要性投影值≥1、差异倍数对数值绝对值≥1 为筛选标准,筛选差异特征挥发性代谢物。
酶活实验层面,分别利用信号肽与跨膜结构域在线预测网站分析角鲨烯合酶蛋白结构;将全长基因及截短序列克隆至原核表达载体,测序验证无误后转化大肠杆菌 BL21 (DE3) 菌株诱导异源表达,参照已发表成熟实验体系完成体外酶活测定与催化产物鉴定。
定量统计分析方法
- 基因组组装评估 :以陆生植物 BUSCO 完整基因占比评估组装完整度,Merqury 软件 k-mer 一致性分值评定碱基准确度;
- 系统发育分析 :OrthoFinder 构建进化树,MCMCTree 贝叶斯模型结合化石点估算分化时间,后验概率判定进化分支置信度;
- 共线性与多倍化分析 :PAML 软件 YN00 算法计算同义替换率,校正演化速率偏差后判定全基因组加倍事件;
- 基因家族分析 :CAFE 软件完成基因家族扩张收缩统计,多重检验校正筛选显著差异家族;
- 代谢组差异分析 :严格遵循双阈值筛选差异挥发性物质,依托专业质谱分析软件完成物质定性定量;
- 通用规则 :无特殊说明情况下,所有分析软件均采用默认参数;所有实验重复数、统计指标、检验方法均详细标注于图注与结果章节内。