采访会议纪要
上回放:《Nature》一作采访会议纪要
采访Nature一作:蛋白酶体介导血红素信号轴调控T细胞耗竭
SCI一作面对面访谈 第三期:徐颖茜教授 《Nature》"蛋白酶体介导血红素信号轴调控T细胞耗竭"
参会人员
徐颖茜教授、Biomamba
会议内容
徐教授以第一作者身份在**《Nature》** 发表题为"Proteasome-guided haem signalling axis contributes to T cell exhaustion"的重磅论文,本次她围绕蛋白酶体调控血红素信号轴介导T细胞耗竭这一研究成果展开详细讲解,并在分享结束后与现场观众开展问答交流。
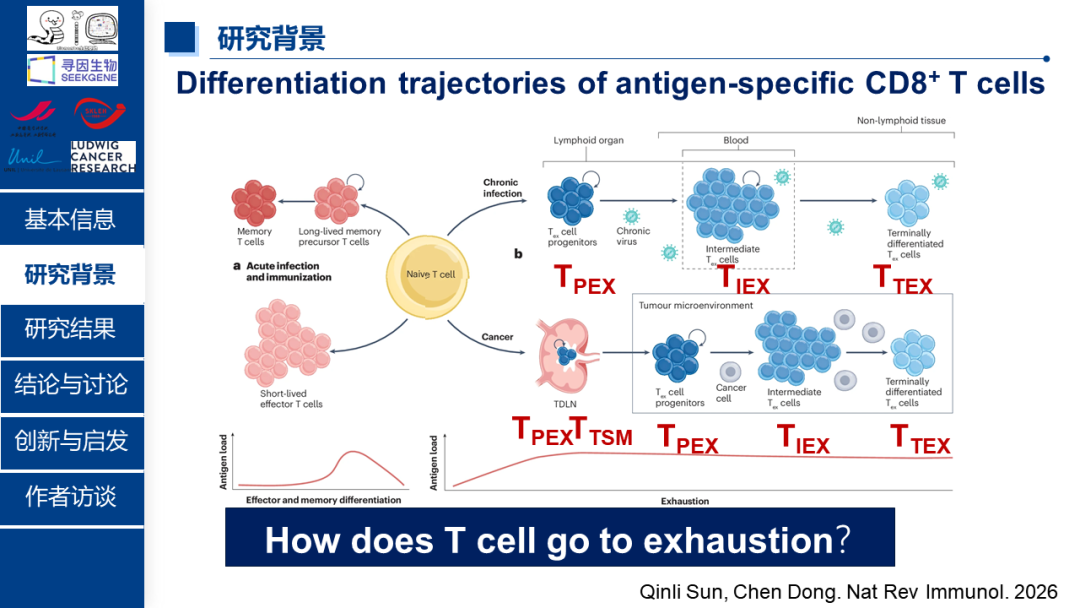
1、研究背景(提出科学问题:How does T cells go to exhausion?)
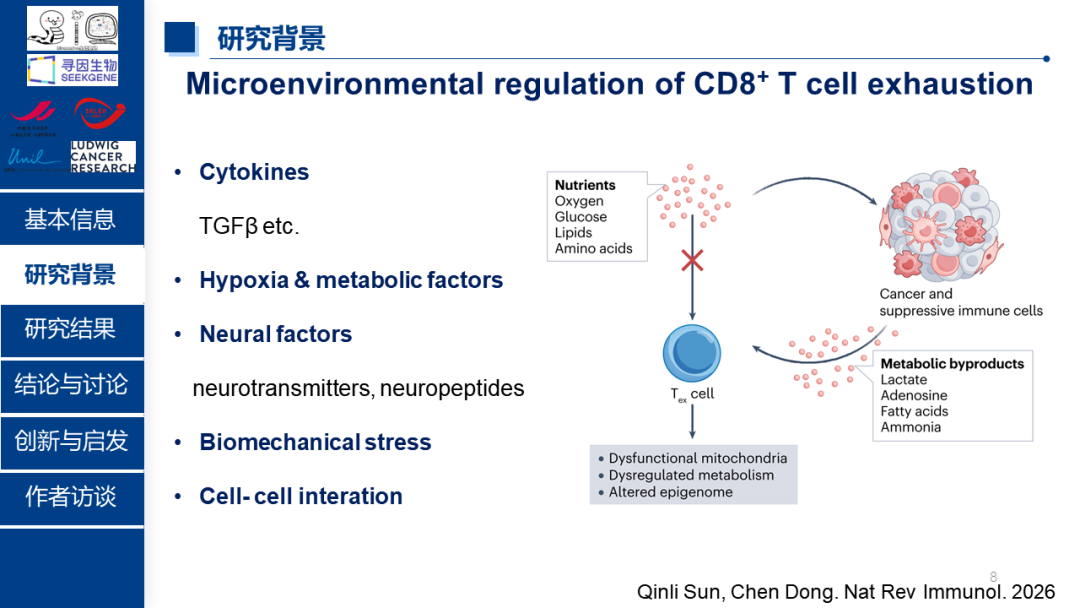
在肿瘤和慢性感染中,CD8⁺ T细胞会逐渐失去杀伤能力,进入一种被称为"耗竭"的状态。耗竭T细胞不仅效应功能下降,增殖能力减弱,还高表达多种抑制性受体。虽然已有研究发现耗竭T细胞中线粒体功能严重受损,但这些"残破"的线粒体如何推动T细胞走向耗竭,其背后的分子机制待解析。
2、研究结果
详细见: 直播采访《Nature》一作:蛋白酶体介导血红素信号轴调控T细胞耗竭
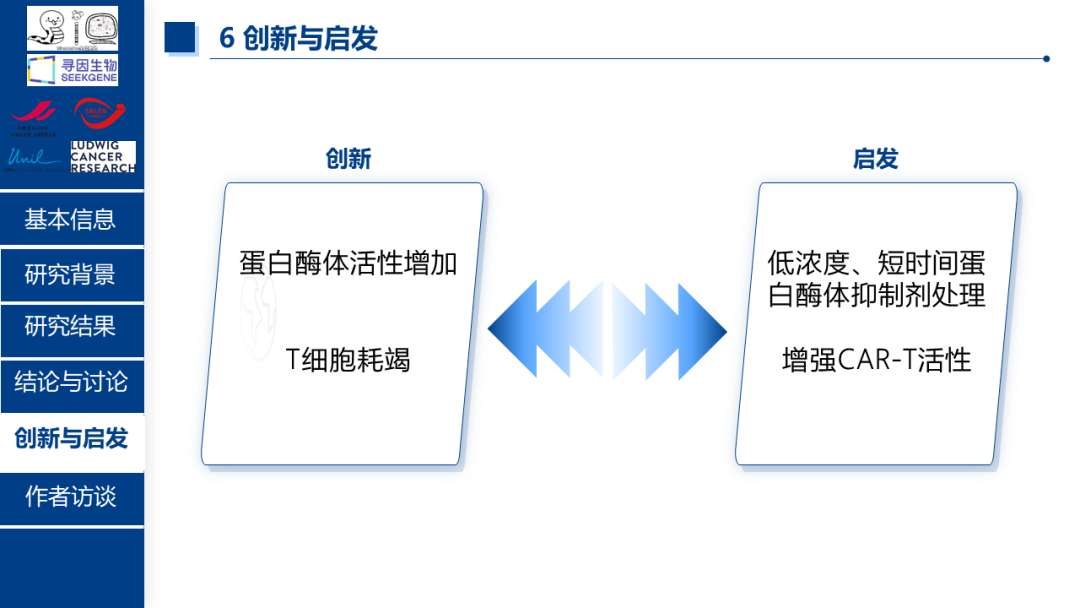
3、创新点与启发:
本研究最核心的创新之处在于建立了蛋白酶体活性 与 T 细胞耗竭之间的调控关联,并阐明线粒体功能紊乱介导 T 细胞耗竭的内在分子机制。上述调控通路为免疫治疗及临床转化提供了关键理论启示:低浓度、短时间干预蛋白酶体抑制剂可显著提升抗体药物的免疫效应,该策略具备广阔的临床转化应用前景。
4、问答环节
①主持人 :科研工作者在撰写标书或在做立项的时候,常常会头疼如何选择关键分子靶点及通路,针对这一棘手问题,本研究为何聚焦**heme对T细胞耗竭的调控?**是参考血红素在其它细胞当中的调控报道或者是通过本课题组的预实验研究等确定的呢?
徐教授: 目前,关于血红素(heme)调控T细胞功能的研究尚未见报道。我们的研究设想来源于既往文献中关于B细胞的研究发现:血红素能够调控BACH2-BLIMP1信号轴,从而影响B细胞 的分化过程。已有大量研究表明,BACH2 和BLIMP1是调控T细胞耗竭 的关键转录因子,在T细胞命运决定和功能维持过程中发挥重要作用。因此,我们提出这样一个科学假设:**血红素是否也能够通过调控BACH2-BLIMP1信号通路参与T细胞耗竭的发生与发展过程?**基于这一假设,我们进一步探索血红素在T细胞耗竭调控中的潜在作用及其分子机制。
②主持人 :在确定血红素对T Cells耗竭调控后,在反向证明诱导血红素降解可以增强T细胞活性,该论文做了怎样的工作和努力呢?
**徐教授:**本研究采用了多种策略评估降低细胞内heme水平与T cell耗竭的影响。
**策略一:抑制heme合成,**heme不仅参与T细胞耗竭调控,同时也是线粒体电子传递链的关键辅因子。实验结果表明,在T细胞进入耗竭状态之前,抑制heme合成会显著削弱其增殖能力和效应功能。
策略二:通过HO-1促进heme降解。结果显示,heme降解不仅会影响ETC功能,抑制T细胞增殖和效应功能,还会释放游离铁离子,进一步诱导T细胞发生铁死亡。
**策略三:增强heme外排。**通过过表达heme转运蛋白促进heme外排。检测发现,肿瘤微环境中的heme浓度本身已经较高,甚至高于T细胞内水平。在这种浓度梯度下,增强外排难以有效降低细胞内heme含量,因此未能改善T细胞功能。
核心认识:决定作用的不是heme总量,而是heme定位。 上述结果表明,无论是抑制合成、促进降解还是增强外排,单纯降低heme总量均无法有效提升T细胞活性。这提示我们,heme在T细胞中的生物学作用可能更多取决于其亚细胞分布,而非总体含量。**关键发现:阻断heme入核,抑制T细胞耗竭。**基于这一认识,本研究将研究重点从"调控heme水平"转向"调控heme定位"。最终发现,阻断heme进入细胞核、避免其对BACH2信号轴的调控,能够在保留heme代谢功能的同时有效抑制T细胞耗竭。
③ 主持人问:作为一个中间介导的分子,过表达Bach2可以在哪些方面增强CAR-T抗肿瘤活性的呢?
**徐教授:关键发现:BACH2持续高表达并不能进一步提升CAR-T疗效。**针对BACH2是否可以增强CAR-T抗肿瘤活性的问题,最初假设:既然BACH2能够抑制T细胞耗竭,那么持续提高其表达水平可能增强CAR-T功能,并据此构建了不可降解的突变体BACH2以维持其持续高表达状态。BACH2在维持T细胞功能与记忆性方面具有重要作用。然而,我们的研究结果显示,当BACH2处于持续高水平、尤其是无法正常降解的状态时,并不能进一步增强CAR-T细胞的抗肿瘤效果。
**核心结论:CAR-T功能依赖于"干性与效应分化"的动态平衡。**其机制在于,BACH2的核心功能是维持T细胞干性并促进memory-like状态,同时抑制其向终末效应T细胞分化。适度维持干性有助于T细胞长期存活与持续反应,但过强的BACH2活性会使T细胞长期停留在记忆样状态,限制其向具有杀伤功能的effector T cells分化,从而削弱实际抗肿瘤效应。因此,BACH2的作用并非"越高越好",而是需要维持在一个动态平衡区间,才能实现最佳的抗肿瘤效果。
④主持人问:终末耗竭T细胞会特异性诱导线粒体蛋白降解是发生原因还是结果?
**徐教授:**我个人倾向于认为,终末耗竭T细胞中出现的线粒体蛋白特异性降解,更多是一种"结果",而不是最初的"原因"。
当T细胞进入终末耗竭状态后,线粒体功能已经明显受损。**蛋白损伤触发细胞质控机制:**线粒体功能障碍会导致活性氧(ROS)大量积累,而过量ROS会引起线粒体蛋白氧化损伤以及错误折叠。为了维持细胞内蛋白质稳态,细胞会选择性地清除这些受损蛋白,因此我们观察到了线粒体蛋白的特异性降解现象。受损蛋白首先被泛素化,随后被蛋白酶体识别并降解。而在这一过程中,E3泛素连接酶往往发挥决定性的作用,是整个泛素化过程的限速步骤。实际上,我们分析了一系列与T细胞耗竭相关的E3连接酶,发现CBLB可能参与介导部分线粒体蛋白的特异性泛素化,从而促进其降解。
因此,我认为一个非常值得关注的问题是:不同的E3连接酶可以选择性识别不同类型的线粒体蛋白,并驱动不同的降解程序。那么未来通过靶向特定E3连接酶来调控线粒体蛋白稳态,进而改善T细胞耗竭状态,可能会成为潜力研究方向。
⑤主持人问:为何用scATAC证明Bach2靶向基因通路及调控T细胞耗竭的分子机制? 针对文章返修意见补了scATAC-seq实验,用到的是我们此前分享过的SeekOne-DD单细胞ATAC+RNA一胞双组学的技术。为何用scATAC证明Bach2靶向基因通路及调控T细胞耗竭的分子机制?
徐教授: 研究中发现,短时间、低剂量的硼替佐米处理后,CAR-T细胞能够获得长期的功能增强效应。审稿人提出:为什么一个短暂的处理,能够产生如此持久的生物学效应?
仅仅从基因表达层面可能无法给出这个问题的完整解释。目前普遍认为,表观遗传重塑(epigenetic reprogramming)是决定T细胞命运的重要机制。一旦T细胞形成特定的表观遗传状态, 其分化方向与功能状态便可能被长期锁定。因此,我们通过整合ATAC-seq与RNA-seq,系统评估硼替佐米处理是否重塑了T细胞耗竭相关的表观遗传程序,以此阐明其长期效应的分子基础。 选择单细胞技术而非传统Bulk测序,关键在于CAR-T细胞群体的高度异质性。无论在体外扩增阶段还是回输患者体内后,CAR-T 群体中均并存处于不同分化阶段与功能状态的细胞亚群。Bulk测序所得仅为群体平均信号,关键亚群的特征极易被稀释掩盖,从而制约机制解析的深度与精度。为此,我们采用scATAC-seq与单细胞转录组联合分析,在单细胞分辨率下精细刻画不同细胞亚群的命运轨迹。结果表明,经硼替佐米处理后,T细胞的表观遗传状态与转录特征整体向memory-like T cells偏移,而非走向terminal exhaustion。这一发现为硼替佐米长期增强CAR-T细胞功能提供了关键的机制依据。
主持人: 好的,可以看出scATAC的这个技术对于基因的表达以及调控还是研究还是非常有利的。那么对于这个single-cell ATAC的分析流程:4.6w字长文| SeekOne-DD单细胞ATAC+RNA一胞双组学-学习手册,感兴趣的小伙伴也可以去联系我们的客服微信【Biomamba_zhushou】领取学习的手册。
⑥观众提问:蛋白酶体抑制作用会不会比较广谱,可以作用于多种细胞,它不仅作用于这个线粒体相关降解。
徐教授: 这是一个非常好的问题。硼替佐米是一种广谱性蛋白酶体抑制剂,对多种蛋白质的降解均有抑制作用。但我们对硼替佐米的使用仅限于CAR-T细胞体外制备阶段的短暂处理,并非在回输至小鼠或患者体内后再行给药。 因此,我们所观察到的功能增强效应,可以明确归因于CAR-T细胞本身,而非药物对体内其他细胞的影响。另一方面,尽管硼替佐米对多类蛋白质的降解均有抑制,但我们已证明,T细胞走向耗竭的过程中,线粒体蛋白的降解是最为显著的分子事件之一。基于这一发现,我们的核心思路是:在CAR-T细胞接触抗原之前,通过短时、低剂量地抑制蛋白酶体活性,优先保护线粒体蛋白,进而维持线粒体功能的完整性,以此延缓T细胞耗竭的发生。
⑦观众问:在DC胞当中Bach2是否也会有相应的功能?能否follow这篇Nature的前沿思路,做迁移学习与科研。
徐教授: Bach2和BLIMAP1这条信号通路在B cells中有很大的作用,而且最近也发现它能抑制NK细胞的效应功能。这里有一点值得关注:不同免疫细胞亚群往往会调用相同或不同的转录因子网络来调控自身的命运与功能状态。这是一个非常好的想法,可以进行一些文献学习。探讨Bach2-BLIMAP1在DC中的作用,heme也仍可能起到关键调节因素。因为一部分Bach2的降解是由于PI3K对其磷酸化,而磷酸化Bach2才能结合heme进一步的降解。这是一个很好的题,可以进一步地进行研究。
全平台回放,欢迎关注
B站:https://www.bilibili.com/video/BV1QQXZB7EFT
小红书:http://xhslink.com/o/2J9LKORBPh9
抖音:https://v.douyin.com/LGYrpuJ7u_I/
如果需要单细胞数据分析指导 、生信热点全文复现、自测数据个性化分析 辅导、 实验科研服务和常态化实验学习,欢迎联系**Biomamba_zhushou**。
嘉宾简介

**徐颖茜,南开大学博士,现任中国医学科学院血液病医院(中国医学科学院血液学研究所)副研究员,**师从肿瘤免疫代谢领域专家Ping-Chih Ho教授。目前主要从事免疫细胞代谢与细胞免疫治疗的研究工作,专注于探索神经网络、生理节律、新陈代谢如何诱导免疫细胞在白血病微环境中的功能紊乱。致力于提高免疫细胞或工程性免疫细胞针对恶性血液肿瘤的治疗效果。获得了多项国家自然科学基金和天津市自然科学基金的支持,相关研究成果以第一作者/通讯作者发表于Nature(2026)、Cell Stem Cell(2024)、Sci Adv.(2021)、Leukemia(2021)、Blood Cancer J.(2021)、J Hematol Oncol.(2019、2018)等国际权威期刊,以第一作者/通讯作者于Trends Endocrinol Metab.(2025)、Sci Bull.(2025)、Trends Cancer(2024)、Cancer Immunol Res.(2023)等期刊发表综述6篇、获得嵌合抗原受体相关研究的国家授权专利7项。作为主要完成人获天津市自然科技进步奖2项(2020、2025)。
2026年3月18日,徐颖茜教授 以第一作者身份在《Nature 》(IF=48.5 )发表题为**"Proteasome-guided haem signalling axis contributes to T cell exhaustion"** 的研究论文,详细介绍见:直播采访《Nature》一作:蛋白酶体介导血红素信号轴调控T细胞耗竭。
本文揭示了衰竭的CD8 T Cell由于去极化线粒体的累积增加了蛋白酶体的活性,从而导致线粒体蛋白选择性降解,并通过血红素蛋白分解释放出调节性血红素,进一步破坏了BACH2介导的转录调控,加剧了T Cell的衰竭并损害了其干细胞特性。抑制调节性血红素核进入,可防止BACH2降解,并增强肿瘤特异性 CAR T疗法的疗效。bortezomib(硼替佐米)作为一种获得FDA批准的蛋白酶体抑制剂,可以防止T Cell衰竭并提升CAR-T的治疗效果。
文章摘要
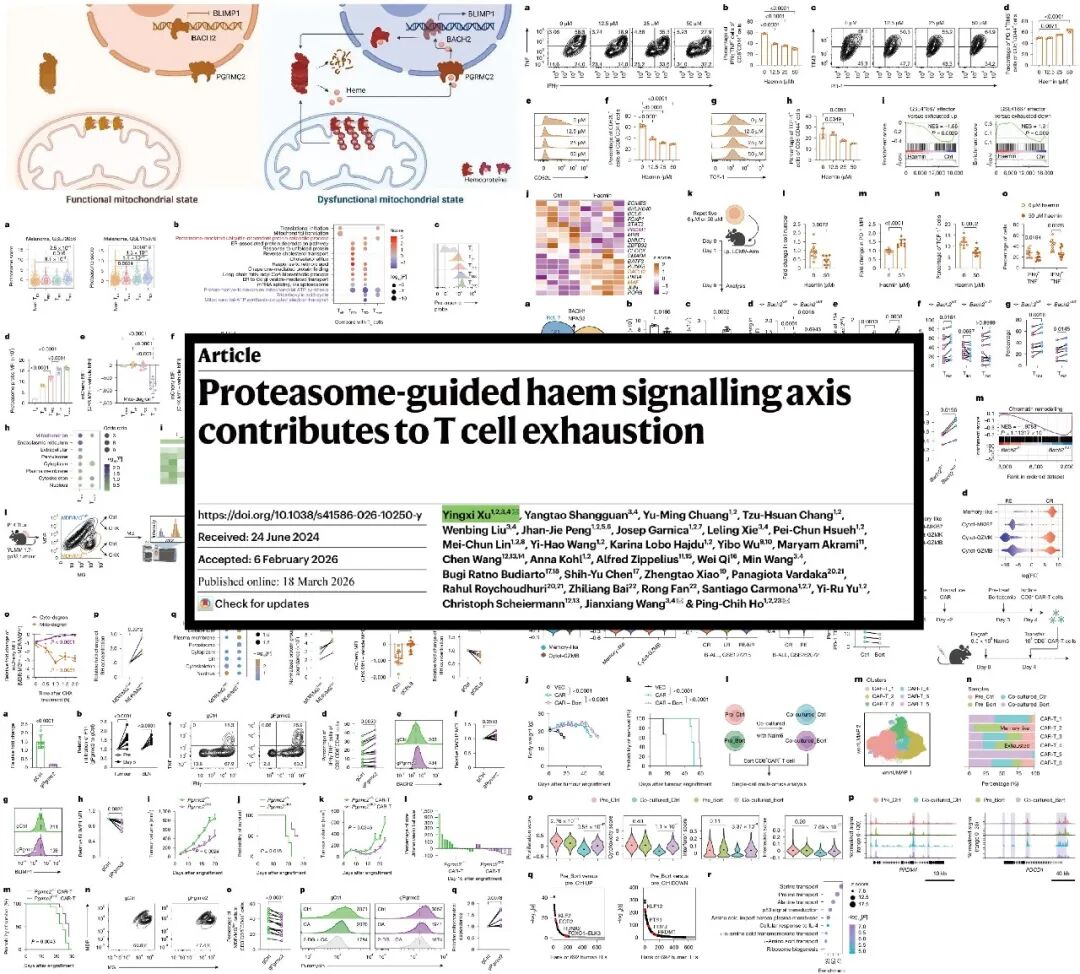
背景:T细胞终末耗竭被认为是限制T细胞免疫疗法获得长期疗效的关键因素之一。处于终末耗竭状态的T细胞通常表现为效应功能下降、增殖能力减弱并持续高表达多种抑制性受体。近年研究表明,细胞代谢在决定T细胞命运中发挥重要作用。其中,线粒体作为细胞能量代谢和信号调控的核心细胞器,其功能异常可驱动T细胞转录重编程并促进其耗竭。然而,去极化线粒体在细胞内的积累如何进一步推动T细胞耗竭并削弱其干性潜能,具体分子机制仍有待深入阐明。
方法 :对耗竭T细胞亚群进行转录组测序和蛋白质组学分析,筛选差异表达基因和蛋白质,并结合黑色素瘤和肺癌患者数据,验证蛋白酶体活性与耗竭的相关性。研究团队敲除了CBLB和PGRMC2等基因,构建了BACH2血红素结合位点突变体,以探究潜在的分子机制。他们还用硼替佐米(蛋白酶体抑制剂)处理CAR-T细胞,并在患有白血病的**C-NKG小鼠(重度免疫缺陷小鼠)**上验证CAR-T细胞的抗肿瘤效果。
结果:研究团队通过蛋白质组学分析发现,在含有去极化线粒体的T细胞中,蛋白酶体活性显著增强。进一步的泛素化蛋白质组分析表明,线粒体蛋白的泛素化水平明显升高,并被蛋白酶体选择性降解,其中包括大量含血红素的电子传递链复合物,而含血红素蛋白的降解可增加去极化线粒体T细胞中调节型血红素(RH)的水平。功能研究显示,升高的RH可促进耗竭标志分子PD-1和TIM3表达,同时削弱T细胞分泌TNF和IFNγ等效应细胞因子的能力。结合RNA-seq和ATAC-seq整合分析发现,RH通过抑制T细胞干性转录因子BACH2的功能,增强终末耗竭相关转录因子BLIMP1的转录活性。机制研究进一步表明,突变BACH2的血红素结合位点可显著抑制T细胞的终末耗竭分化,而抑制血红素入核则可增强T细胞的抗肿瘤功能。
结论 :这项研究揭示了蛋白酶体-血红素-BACH2信号轴调控CD8+ T细胞耗竭的新机制。 去极化线粒体的积累导致蛋白酶体活性增强,在此过程中,蛋白酶体选择性降解线粒体蛋白来维持细胞稳态,并导致调节型血红素的释放。血红素水平升高通过调节BACH2调控的转录网络来强化表型和转录重编程,从而驱动T细胞耗竭。这些结果表明,以蛋白酶体活性和血红素核转运为靶点,有望提高CAR-T细胞治疗的效果和持久性。