
Decoding enzymatic landscapes: a knowledge graph--enhanced large language model framework for microbial enzyme production and catalysis systems
01
摘要
研究团队构建了MEPAM问答系统,用三个机器学习模型从7万余篇文献中筛出1.1万篇高质量论文,借助DeepSeek-V3零样本抽取1.2万实体与3.6万关系构建知识图谱,结合RAG技术实现准确率达0.86、几乎零幻觉的酶催化知识问答。
02
文献海洋中的导航难题
酶,作为自然界最高效的催化剂之一,在工业制造、农业生产和环境治理中扮演着不可替代的角色。无论是生物燃料的制备、食品加工,还是制药中间体的合成,背后都离不开微生物酶的表达与催化。要让一个酶真正"好用",科研人员必须同时搞定两件事:一是找到合适的微生物宿主来实现酶的高效可溶性表达,二是精确调控发酵与催化反应条件以获得最高活性。这两件事涉及的变量极多------宿主菌种、培养基配方、温度、pH、底物浓度、金属离子------它们之间交织成一张复杂的网络。
可问题在于,这张网络至今没有被系统地描绘出来。
在Web of Science数据库中,仅以"微生物"和"酶"为关键词检索,返回的文献数量就超过百万篇。即便缩小到发酵、可溶性表达等更具体的查询,候选文献仍有七万多篇。科研人员面对的不是一个信息匮乏的世界,而是一个信息过载到令人窒息的世界。更要命的是,生命科学领域的术语体系极其混乱。同一种枯草芽孢杆菌,在NCBI数据库中叫Bacillus subtilis 168,到了德国微生物保藏中心则变成了DSM402。大肠杆菌在文献中至少有37种不同写法,枯草芽孢杆菌也有26种。"木糖"这个词,既可能是培养基成分,也可能是酶催化的底物------上下文不同,含义截然不同。
这种混乱直接导致了一个尴尬的局面:即便你想用知识图谱等计算工具来整合已有发现,数据收集本身就成了瓶颈。人工筛选费时费力,覆盖面窄;而直接让大语言模型来回答问题,它又可能一本正经地编造数据。
有没有办法既利用大模型的理解能力,又约束它的"想象力"?这就是这篇发表在aBIOTECH上的研究试图回答的核心问题。
03
三道筛子:从77087到11068
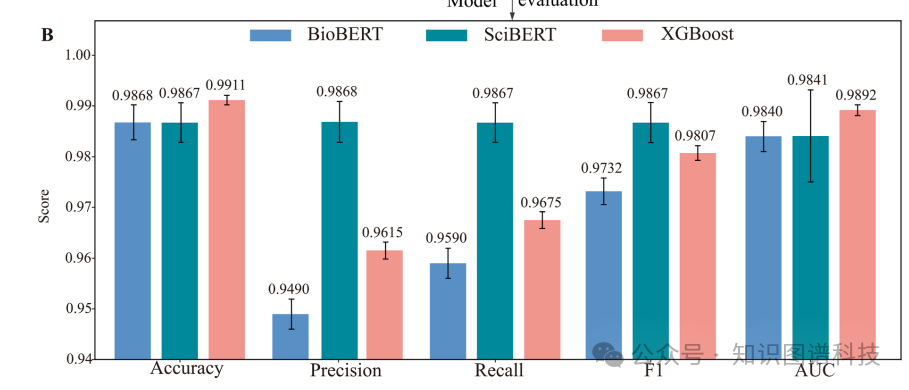
研究团队的第一步,是解决"文献从哪来"的问题。他们设计了一套系统化的检索策略,以发酵、可溶性表达、酶以及五种常见微生物宿主(枯草芽孢杆菌、大肠杆菌、酵母、里氏木霉、曲霉)为关键词组合,在Web of Science中检索2000年至2025年间发表的英文原创研究论文,初筛获得77087篇候选文章。这些文章分布在4356种期刊上,其中影响因子大于10的有2804篇,出现频率最高的包括Applied and Environmental Microbiology(1905篇)、Journal of Biological Chemistry(1891篇)和Journal of Bacteriology(1871篇)。
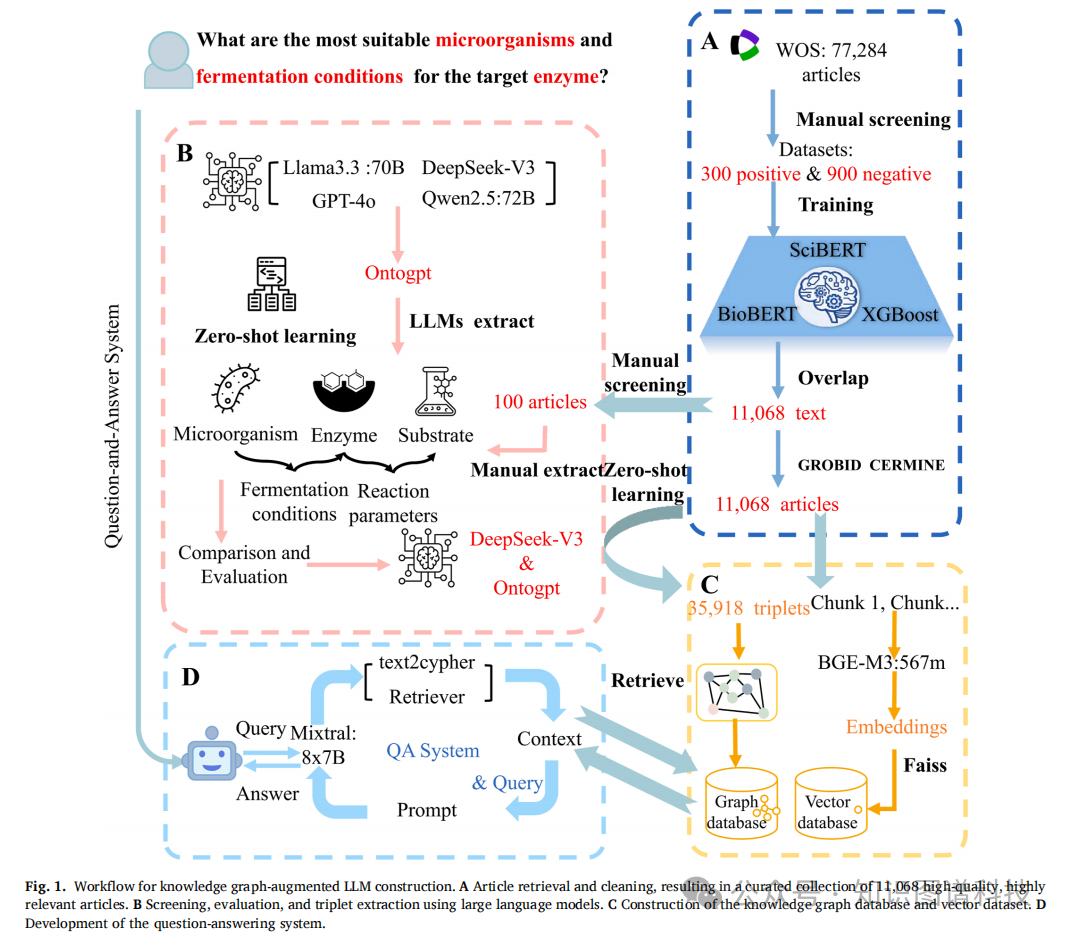
七万多篇文献显然不能直接拿来用。研究团队训练了三个分类模型------BioBERT、SciBERT和XGBoost------来对文献进行相关性筛选。训练集由300篇正面文章和900篇负面文章的摘要构成,三个模型在测试集上的表现都相当出色,准确率、AUC值和F1分数均超过0.98。
将三个模型分别应用于77087篇候选文献后,BioBERT预测出51121篇相关,SciBERT给出40816篇,XGBoost则更保守,仅认定31216篇。研究团队采取了最严格的策略:只保留三个模型一致认定的11068篇文章。这11068篇文献覆盖了88种期刊,其中影响因子大于10的有263篇,出现频率最高的期刊是Applied and Environmental Microbiology(332篇)、Bioresource Technology(279篇)和Applied Microbiology and Biotechnology(198篇)。
这种"三模型共识"策略的好处显而易见:它牺牲了召回率,但最大限度地保证了精度。后续构建知识图谱的每一篇文献,都经过了三个独立模型的交叉验证。
04
大模型选美:DeepSeek-V3凭什么胜出
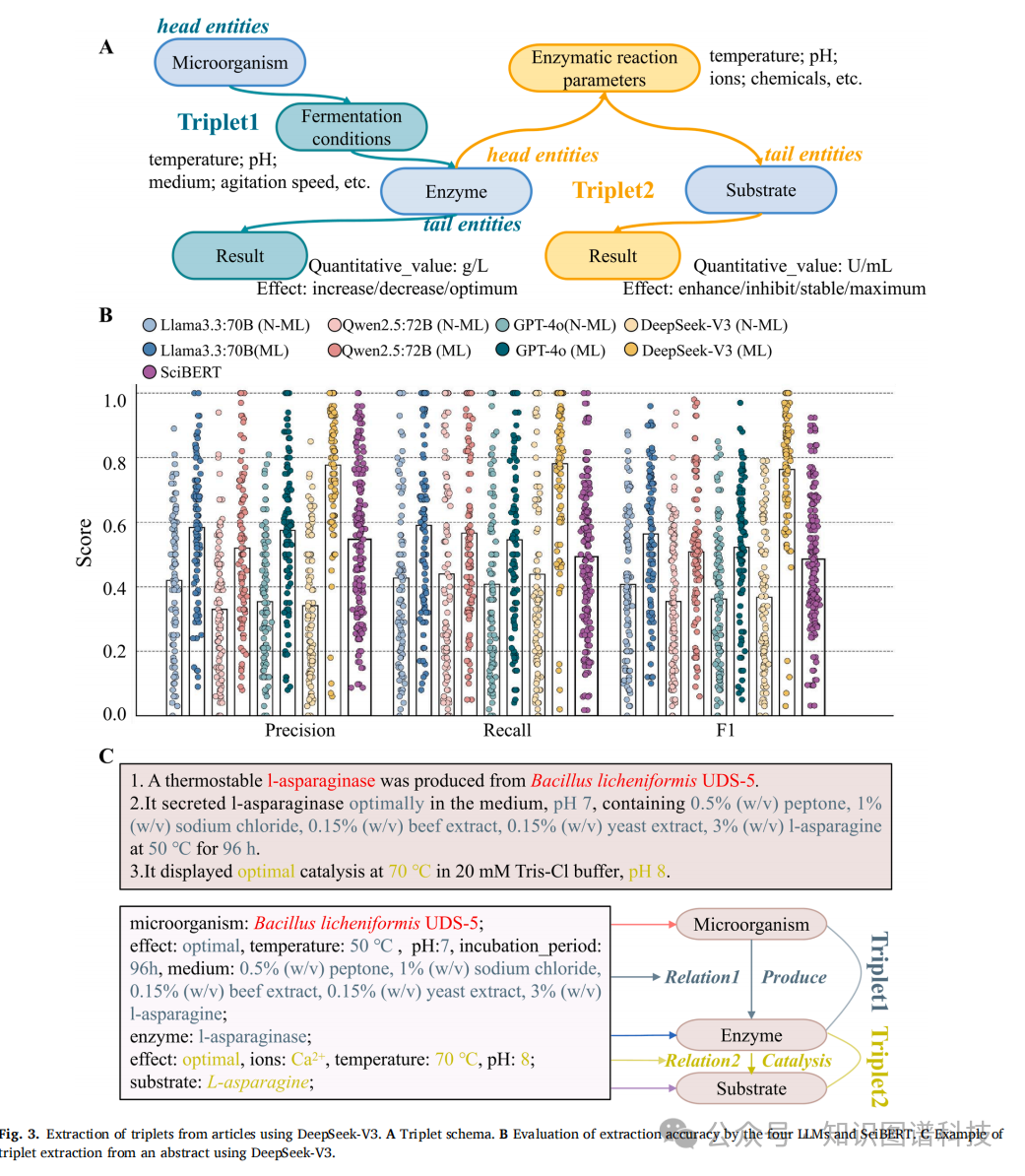
有了高质量语料,下一步就是从中抽取结构化知识。研究团队定义了三个核心实体------微生物、酶、底物------以及两类关系:微生物到酶的"发酵条件"(属性包括pH、温度、培养基组分、碳源、氮源等),和酶到底物的"酶催化参数"(属性包括温度、离子、底物浓度等)。这构成了两种基本三元组结构:(微生物)-发酵条件-(酶),以及(酶)-酶催化参数-(底物)。
为了让大模型准确理解这套Schema,团队借助DSPY框架将实体和关系的定义、约束规则写入YAML配置文件,要求模型在抽取时严格保留原文内容,禁止任何修改、解释或生成性输出。这种"戴着镣铐跳舞"的设计,本质上是用结构化Schema来约束大模型的自由度,从源头防止幻觉。
接下来要回答一个关键问题:用哪个大模型来抽取?
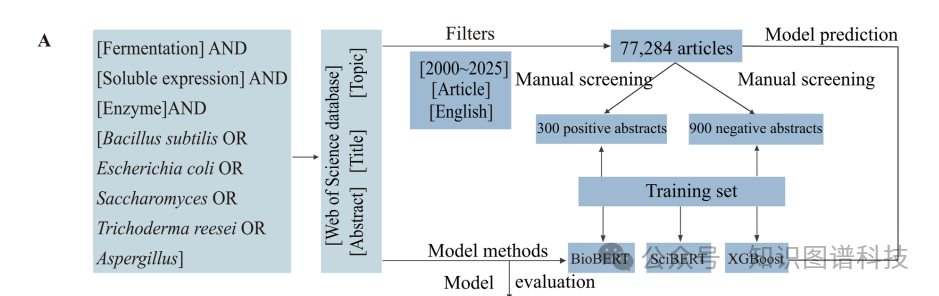
团队选取了四个当时最先进的通用大模型进行横向比较:DeepSeek-V3、Qwen2.5-72B、Llama3.3-70B和GPT-4o。他们从11068篇文章中随机抽取100篇,用BRAT工具手动标注了422条三元组作为标准答案,然后让四个模型分别对这100篇文章进行零样本三元组抽取,再用Qwen2.5-14B作为裁判来打分。
结果很明确。DeepSeek-V3配合LinkML格式的Schema约束,在精确率、召回率和F1分数三项指标上均以0.78、0.78、0.76的成绩领先,其余三个模型的三项指标都徘徊在0.5到0.6之间。更重要的是,所有使用LinkML格式约束的模型,表现都优于不加约束的版本------这说明结构化的Schema提示对抽取质量至关重要。与微调过的SciBERT相比,DeepSeek-V3在三项指标上分别提升了41.8%、59.2%和55.1%。
但精度只是考量的一个维度。研究团队还算了一笔经济账。按照2025年4月的公开价格,DeepSeek-V3每百万tokens仅售0.5元(64K上下文),是四个模型中最便宜的;GPT-4o则高达18.1元,贵了36倍。处理速度方面,DeepSeek-V3处理100篇论文仅需0.42小时,GPT-4o需要0.83小时,Qwen2.5-72B最慢,要1.92小时。
精度最高、成本最低、速度最快------DeepSeek-V3几乎以压倒性优势赢得了这张"合同"。随后,研究团队用它对全部11068篇文章进行了大规模三元组抽取。在此之前,PDF文件先经过GROBID和CERMINE两个工具转换为结构化XML格式,摘要部分的解析准确率达到0.94,结果部分为0.89,保证了文本挖掘的输入质量。
最终,整条流水线共抽取了12434个实体,包括1842种微生物、7310种酶和3282种底物,以及35918条关系,其中发酵条件19386条、酶催化条件16532条。
举一个具体的抽取案例。论文摘要中写道:"一种耐热L-天冬酰胺酶由地衣芽孢杆菌UDS-5产生;该酶在pH 7、含0.5%蛋白胨、1%氯化钠、0.15%牛肉膏、0.15%酵母膏、3% L-天冬酰胺的培养基中,50摄氏度培养96小时,分泌量最优;在70摄氏度、20 mM Tris-Cl缓冲液、pH 8条件下催化活性最优。"DeepSeek-V3成功从中拆解出完整的结构化信息:微生物为地衣芽孢杆菌UDS-5,发酵条件包括最适温度50摄氏度、pH 7、培养时间96小时及具体培养基配方,酶为L-天冬酰胺酶,催化条件包括最适温度70摄氏度、pH 8、Ca2+离子,底物为L-天冬酰胺。这种从自由文本到结构化三元组的精确转化,正是整条知识图谱构建流水线的基本单元。
05
知识图谱里藏着什么
有了上万个实体和近四万条关系,研究团队开始对这张知识图谱进行系统分析。
微生物实体涵盖了170个属、1842个种。出现频率最高的是酵母属(Saccharomyces,32.0%),其次是大肠杆菌属(Escherichia,28.0%)和木霉属(Trichoderma,12.1%)。到了种一级,优势物种更加集中:酿酒酵母占了酵母属的95.7%,大肠杆菌几乎包揽了大肠杆菌属的全部,里氏木霉则占木霉属的96.0%。这三种微生物显然是酶生产领域最热门的宿主选择。
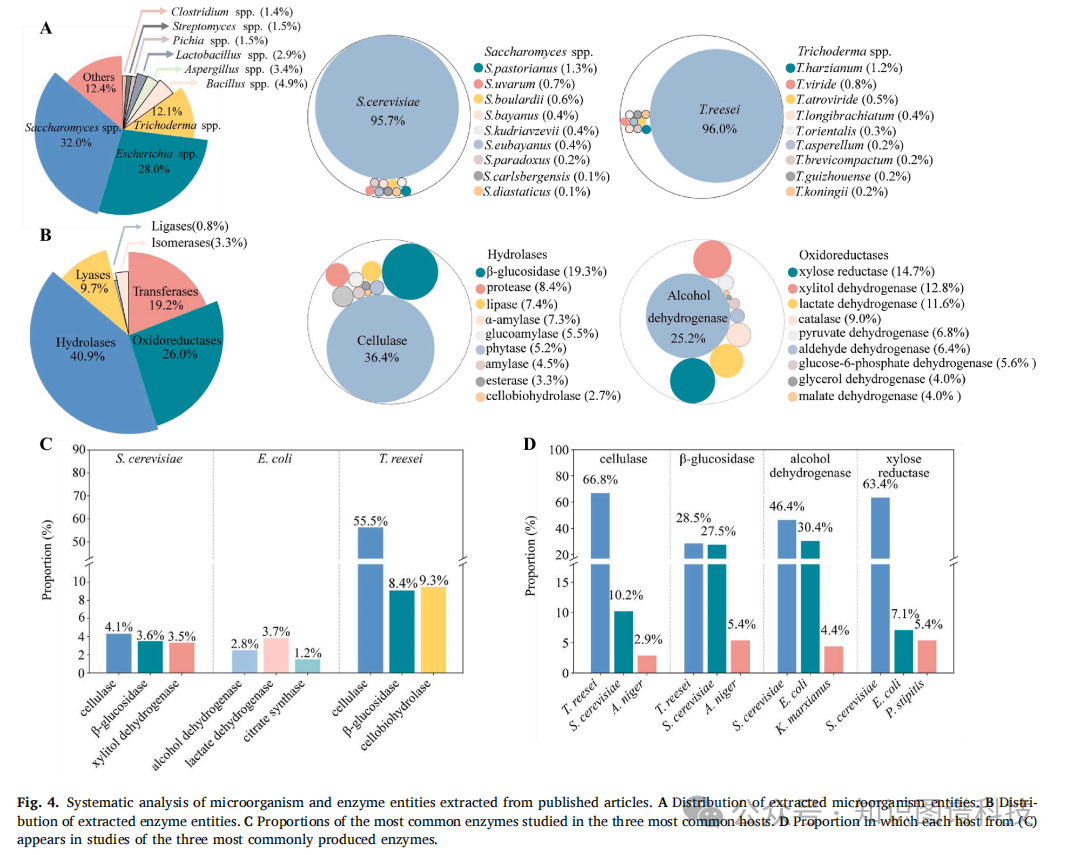
7310种酶被归入六大功能类别。水解酶占比最高,达40.9%,其次是氧化还原酶26.0%。在水解酶中,纤维素酶占36.4%,β-葡萄糖苷酶占19.3%;氧化还原酶中,醇脱氢酶占25.2%,木糖还原酶占14.7%。
如果把酶和宿主交叉来看,一些有趣的规律浮出水面。里氏木霉是纤维素酶的绝对主力,66.8%的纤维素酶由它生产,其中里氏木霉自身的产酶谱中纤维素酶占55.5%。酿酒酵母则更偏向醇脱氢酶(46.4%)和木糖还原酶(63.4%),这与它在生物乙醇发酵中的核心地位高度吻合。大肠杆菌产的酶种类最为分散,每种酶占比在0.05%到3.67%之间------这大概得益于它遗传背景清晰、可操作性强、培养成本低,什么酶都能拿来试试。
这些数据背后,隐约可见一条产业主线:将纤维素类农业废弃物(玉米秸秆、甘蔗渣等)转化为可发酵糖,再用于生物乙醇生产。纤维素酶和β-葡萄糖苷酶在里氏木霉和酿酒酵母中的研究,正是围绕这条路径展开的。
06
以纤维素酶为例:一张网络的全景图
为了展示知识图谱的实际分析能力,研究团队以里氏木霉产纤维素酶为例,做了深入的网络解析。
从19386条发酵条件数据中,可以清晰地看到里氏木霉产纤维素酶的典型工艺参数:温度集中在30摄氏度左右,pH偏弱酸性,通常在4.5到6之间,最常见的是5。也有不少文献报道了28摄氏度和50摄氏度的发酵温度。碳源以纤维素、乳糖、葡萄糖和微晶纤维素Avicel为主,氮源则常用硫酸铵和尿素。发酵周期多集中在48、72、120、144和168小时,搅拌速度从120到500转每分钟不等,通气量在0.3到4 vvm之间。文献中报道的纤维素酶最高产量达100克每升,最高酶活为296 U/(mL·min)。

催化反应方面,研究团队分析了纤维素酶对四种底物的水解条件:木质纤维素、纤维素、羧甲基纤维素(CMC)和Avicel。纤维素是最受青睐的底物,相关实验数据最多。纤维素酶水解纤维素的最适条件为30到50摄氏度、pH 5,呈现出中温弱酸性的特征。木质纤维素和CMC的规律类似。而Avicel的水解出现了30、50、60和80摄氏度多个最适温度点,暗示存在耐高温酶活的变体。
不过,研究团队也注意到一个普遍问题:大部分文献只报告了相对改善(比如"产量提高"或"活性增强"),缺少绝对的酶产量或酶活数据。这导致很难跨研究比较工业生产水平的真实状况。为此,他们在关系抽取中引入了"效应"参数------发酵条件分为增加、降低、最优三类,催化参数分为增强、抑制、稳定、最大四类------只保留包含这些效应词的数据条目,确保分析结果有实际参考价值。
07
MEPAM:一个能溯源、不幻觉的问答系统
知识图谱建好了,但它的价值最终要体现在"能不能回答问题"上。研究团队基于检索增强生成(RAG)技术,开发了MEPAM问答系统。
MEPAM的工作流程分三步走。当用户输入一个问题时,系统首先用Mistral 8x7B模型提取关键词,同时从向量数据库中检索语义最相关的前5条文档片段。接着,提取的关键词通过text2cypher模块转换为Cypher查询语句,在NebulaGraph图数据库中检索关联的实体和关系。最后,图数据库返回的结构化数据和向量检索结果连同原始来源一起,作为上下文喂给Mistral 8x7B,生成最终回答。
这个设计的巧妙之处在于"双通道检索":向量检索负责模糊语义匹配,图数据库负责精确结构化查询,两者互补。每一条回答都可以追溯到原始论文的DOI,用户能直接查证。系统检索语义相似度最高的前5条文档片段,同时通过Cypher查询在图数据库中遍历关联的实体与关系,两路结果合并后送入大模型生成最终回答。
效果如何?研究团队构建了一个包含30个专业问题的基准测试集,问题涵盖β-葡萄糖醛酸酶、天冬氨酸转氨甲酰酶、漆酶、纤维素酶等多种酶的动力学参数(如Km、Vmax)、最适反应条件(pH、温度)、底物特异性和抑制剂/激活剂。每个问题都追溯至原始论文并记录DOI,手动提取关键数据作为标准答案。为确保评估公平,所有模型的原始输出都经过统一后处理:人工逐条审核,精确提取与标准答案对应维度的数值、单位或条目,统一格式(如温度统一为摄氏度,离子写为Zn2+形式)。MEPAM在精确匹配、精确率、召回率、F1分数和忠实度五项指标上分别取得0.73、0.84、0.85、0.84和0.93的成绩,全面碾压包括GPT-4o在内的所有基线模型。GPT-4o对应的五项分数仅为0.37、0.55、0.55、0.54和0.67。
最关键的是幻觉率。MEPAM几乎不产生幻觉,而GPT-4o的幻觉率达0.33,DeepSeek-V3、Qwen2.5-72B和Mixtral 8x7B更是超过0.50。作为代价,MEPAM的响应时间约为5秒,比其他大模型的约1秒稍慢------但考虑到准确性的巨大差距,这点等待完全值得。
举一个具体例子。当用户问"黑曲霉或里氏木霉产纤维素酶的最适发酵pH是多少"时,MEPAM通过text2cypher将问题转化为Cypher查询,图数据库返回pH值为4.0和5.0,结合向量检索结果,最终给出精确回答:"4.0和5.0"。而未经知识图谱增强的Mistral 8x7B给出的答案则含糊得多,声称最适pH在4.5到6.0之间。当检索结果不足以完整回答时,MEPAM会透明地标注"此信息可能不完整,包含大模型补充内容",让用户清楚区分哪些来自文献、哪些来自模型推理。
08
为什么选择零样本
一个值得讨论的技术选择是:研究团队最终选择了零样本学习,而非在生物信息领域更常见的微调或少样本方法。
这个决定并非拍脑袋。团队在预实验中发现,少样本学习存在一个隐蔽的陷阱:当示例中出现"最适生长温度37摄氏度"这样的信息时,部分模型会在后续抽取中反复"复读"这个参数,即便原文根本不支持这一说法。换句话说,示例本身可能成为幻觉的源头。而零样本学习通过Schema调整就能快速构建知识图谱,具有更广的泛化性和迁移性,同时也有助于抑制幻觉。
当然,零样本并非没有代价。由于生物学语言的复杂性,同一物种的不同写法会造成抽取遗漏,这在一定程度上拉低了精确率。前文提到的大肠杆菌37种写法和枯草芽孢杆菌26种写法,在手动标注和模型抽取之间制造了不小的偏差。研究团队用64K的长上下文窗口,将每篇文章的完整摘要和结果部分都纳入上下文,尽量让模型从充分的语义信息中做实体消歧。但命名不一致的根本性问题,仍有赖于更完善的参考数据库来解决。这也是为什么抽取准确率停在0.78而非更高------剩下的0.22,很大一部分要归咎于术语混乱本身。
09
从一个领域到一种范式
MEPAM的意义并不局限于微生物酶领域。它实际上展示了一种可复制的范式:用机器学习筛选高质量文献,用大模型零样本抽取结构化知识,用知识图谱存储和查询,用RAG实现可溯源的低幻觉问答。这套框架的每一个环节都是模块化的------换一个领域,只需要调整Schema定义、检索策略和评估指标,就能快速构建该领域的智能问答系统。
对于工业生物技术从业者而言,MEPAM提供了一个可以直接使用的工具。当你想知道某种酶在哪种宿主里表达量最高、发酵条件怎么调、催化最适pH是多少,不用再在七万篇文献里大海捞针,直接问MEPAM即可,每个答案都附有文献溯源,可以直接追溯原始论文验证。对于投资人而言,这套框架揭示了一个信号:知识图谱增强大模型在垂直科学领域的落地,已经从概念验证走到了实用工具阶段,距离商业化应用并不遥远。对于研究者而言,MEPAM的开放代码和公开数据意味着你可以复现整条流水线,甚至把它迁移到自己的研究领域------无论是材料科学、药物发现还是合成生物学,只要存在大量已发表文献和明确的知识结构,这套范式就能落地生根。
当然,局限也是存在的。文献中绝对酶活数据的匮乏,限制了跨研究定量比较的深度;术语不一致导致的部分抽取遗漏,仍需更完善的实体消歧策略来弥补;5秒的响应时间在实时交互场景中也有优化空间。研究团队也坦诚指出,当前数据集主要来自英文文献,非英语研究社区的成果尚未纳入,这在覆盖面上存在一定偏差。但瑕不掩瑜,当一个大模型能够在专业领域做到0.86的准确率和几乎为零的幻觉率时,它已经从一个"聊天工具"变成了一个值得信赖的研究助手。