今日解读聚焦宏组学与病毒生态调控领域近期发表于顶刊的4篇核心研究。土壤病毒组研究发现病毒编码固氮基因nifU,可提升土壤固氮活性。医学宏基因组研究在胃癌患者的口腔-肠道轴中,鉴定出源自口腔的特定乳酸菌标志物,并构建了高精度诊断模型。农业宏转录组研究锁定毛螺菌科是调控奶牛瘤胃生物氢化和乳汁ω-6/ω-3多不饱和脂肪酸比例的关键类群。环境微生物组研究通过绝对定量,发现家庭灰尘中微生物的绝对丰度与居民健康症状相关,并首次揭示胞外囊泡是重要的毒力因子载体。研究从家禽抗感染机制、鼻咽癌精准筛查、当归根腐病绿色防控,到蜱传耐药基因生态风险,系统揭示宏转录、宏病毒与微生物组在健康、农业及公共卫生中的关键作用,为疾病防治、病害绿色管控与耐药性防控提供新靶点与新思路。
● 文章1 Nature Communications(IF=15.7)
中国科学院生态环境研究中心------根际病毒在促进土壤固氮中的隐藏作用
● 文章2 Cell Reports Medicine(IF=10.6)
上海交通大学医学院附属仁济医院------胃癌患者肠道和口腔微生物群的特征
● 文章3 Microbiome (IF=12.7)
南京农业大学动物科学院------毛螺科是调节奶牛瘤胃生物氢化和乳中ω-6/ω-3多聚脂肪酸比例的关键类群
● 文章4 Microbiome (IF=12.7)
厦门市室内空气与健康重点实验室,区域与城市生态国家重点实验室,中国科学院城市环境研究所------揭示中国家居粉尘微生物风险:从绝对丰度到毒力单位的综合分析
● 文章5 Nature Communications(IF=15.7)
吉林农业大学动物医学院------盲肠Intestinimonas抑制E. tenella配子发育、增强免疫抗感染
● 文章6 Gut(IF=26.2)
中山大学肿瘤防治中心------肠道微生物组用于EB病毒相关鼻咽癌的准确诊断和预后预测
● 文章7 Microbiome(IF=12.7)
南京农业大学资源与环境科学学院------解析中药当归根际微生物组组装机制,揭示代谢物驱动的合生素防病策略
● 文章8 npj Biofilms and Microbiomes(IF=9.2)
第四军医大学公共卫生学院,教育部特殊作业环境危害评估与防护重点实验室,陕西省环境危害评估与防护重点实验室------蜱媒介的抗生素抗性基因动员:跨野生动物-牲畜-媒介-环境界面的跨界传播
文章1 Nature Communications(IF=15.7)
中国科学院生态环境研究中心

研究背景
生物固氮是陆地氮循环的核心,传统上归因于细菌的固氮酶活性。病毒作为地球上最丰富的生物实体,可通过水平基因转移和辅助代谢基因(AMGs)调控宿主代谢,影响碳、磷等元素循环。然而,病毒是否以及如何参与土壤生物固氮过程,特别是是否编码固氮相关基因,尚属未知。
实验设计
**全球数据库挖掘:**从公共病毒组数据库(IMG/VR)中筛选携带固氮基因(NFGs)的病毒。
**田间组学分析:**对南京长期种植豇豆的根际土壤和本体土壤进行病毒组和宏转录组测序,分析病毒AMGs(特别是nifU)的丰度与表达。
**功能验证实验:**通过病毒移植微宇宙实验,结合 ¹⁵N₂稳定同位素标记和DNA稳定同位素探针技术,验证添加根际病毒对土壤固氮活性和微生物群落的影响。
主要研究结果
**1、全球分布:**携带NFGs的病毒在全球病毒组中占比极低(~0.03%),但其分布具有显著地理格局,主要集中于北美、欧洲和亚洲的土壤环境。
**2、关键AMG发现:**鉴定出nifU为病毒编码的固氮相关AMG。该基因在豇豆根际土壤中的丰度和表达水平均显著高于本体土壤。
**3、功能增强证据:**病毒移植实验表明,添加根际病毒悬浮液可使土壤固氮酶活性从1.79显著提升至3.14 nmol C₂H₄ g⁻¹干土 h⁻¹,土壤总氮浓度也同步增加。
**4、微生物群落改变:**病毒添加后,利用¹⁵N标记的DNA-SIP技术发现,活跃固氮的微生物群落中,固氮菌属Azotobacter的相对丰度高达90.8%。
**5、基因来源推测:**系统发育分析显示,病毒nifU序列与细菌同源物具有高度相似性(70-99%),支持其通过水平基因转移从细菌宿主获得。
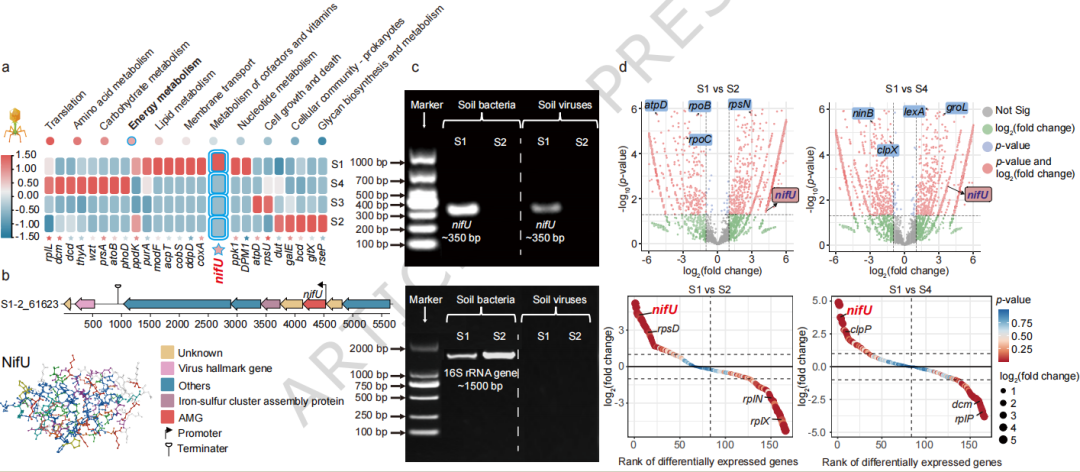
图 豆科和非豆科土壤中病毒AMGs的转录组学验证
文章2 Cell Reports Medicine(IF=10.6)
上海交通大学医学院附属仁济医院

研究背景
胃癌是常见的癌症。除幽门螺杆菌(Hp)外,其他微生物在胃癌中的作用尚不清楚。此前研究发现胃癌患者胃部微生物中富含链球菌和乳杆菌等乳酸菌,但这些细菌是否以及如何从口腔传播至肠道并影响疾病,尚未在口腔-肠道轴上进行系统性研究。
实验设计
**研究对象:**纳入两个独立队列,共317名个体(包括胃癌患者和慢性胃炎对照)。
**样本类型:**收集每位参与者的粪便和唾液样本(队列2)。
**核心技术:**对全部样本进行鸟枪法宏基因组测序,以高分辨率分析微生物组成。
**验证队列:**使用一个独立的哈尔滨队列(舌拭子样本)对口腔标志物进行验证。
**重点分析:**包括物种差异分析、菌株水平追踪、共丰度网络构建和机器学习模型预测。
主要研究结果
**1、发现特异性微生物标志物:**在胃癌患者的肠道中,鉴定出28个差异微生物物种,其中20个是口腔-肠道共享物种,且绝大多数是乳酸菌(如链球菌、乳杆菌)。这些细菌在肠道中富集,但在口腔中并无相同变化。
**2、证实口腔至肠道的菌株传播:**通过对87对匹配的唾液-粪便样本进行菌株水平分析,证实了胃癌相关的链球菌物种(如咽峡炎链球菌)从口腔传播至肠道,支持了"口腔起源"假说。
**3、揭示微生物协同作用:**这些在胃癌中富集的乳酸菌在口腔和肠道微生物组中均形成了紧密的共丰度网络,提示它们可能存在功能协同。
**4、功能通路富集:**胃癌患者粪便中乳酸发酵代谢通路显著富集,与富集的乳酸菌功能一致。
**5、构建高精度诊断模型:**基于上述微生物标志物构建的机器学习模型,能有效区分胃癌与慢性胃炎,粪便模型预测曲线下面积(AUROC)为0.85,唾液模型为0.87,显示出非侵入性诊断潜力。
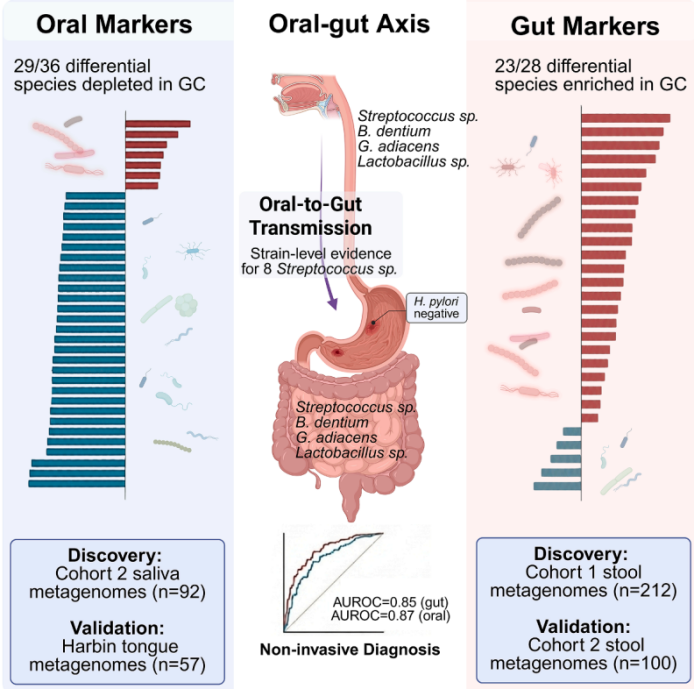
图 图文摘要
文章3 Microbiome (IF=12.7)
南京农业大学动物科学院

研究背景
牛奶中较低的ω-3多不饱和脂肪酸(PUFA)含量和较高的ω-6/ω-3 PUFA比例不利于人类健康。瘤胃微生物的生物氢化过程是决定牛奶脂肪酸组成的关键,但调控牛奶ω-6/ω-3 PUFA比例的具体微生物机制尚不明确。本研究旨在揭示影响该营养指标的关键微生物类群和代谢途径。
实验设计
**动物与分组:**选取95头处于泌乳中期的健康荷斯坦奶牛,根据其乳汁的ω-6/ω-3 PUFA比例,筛选出高比值组和低比值组各15头进行深入分析。
**多组学采样:**收集每头牛的乳汁(用于脂肪酸分析)、瘤胃食糜(饲喂后4小时采集,用于微生物和代谢物分析)和血液样本。
**分析方法:**结合扩增子测序、宏转录组测序和脂肪酸代谢组学,比较两组间微生物群落结构、活性功能及代谢通路的差异,并通过体外培养实验对关键菌株的功能进行验证。
主要研究结果
**1、微生物贡献度:**瘤胃细菌群落结构可以解释奶牛个体间41%的乳汁ω-6/ω-3 PUFA比例差异。
**2、关键微生物类群:**综合多组学分析锁定毛螺菌科是调控PUFA代谢的核心类群。高比值组瘤胃微生物中脂肪酸异构酶基因的转录水平更高。
**3、底物代谢特异性:**体外实验验证,分离到的B. hungatei菌株优先氢化ω-3 PUFA(α-亚麻酸,ALA),而Eubacterium_I菌株则高效代谢ω-6 PUFA(亚油酸,LA)。这种差异化的底物代谢是影响最终乳汁脂肪酸比例的重要机制。
**4、核心结论:**瘤胃微生物(尤其是毛螺菌科)通过差异化的生物氢化作用,调控ALA和LA的代谢流向,从而显著影响乳汁中的ω-6/ω-3 PUFA比例。
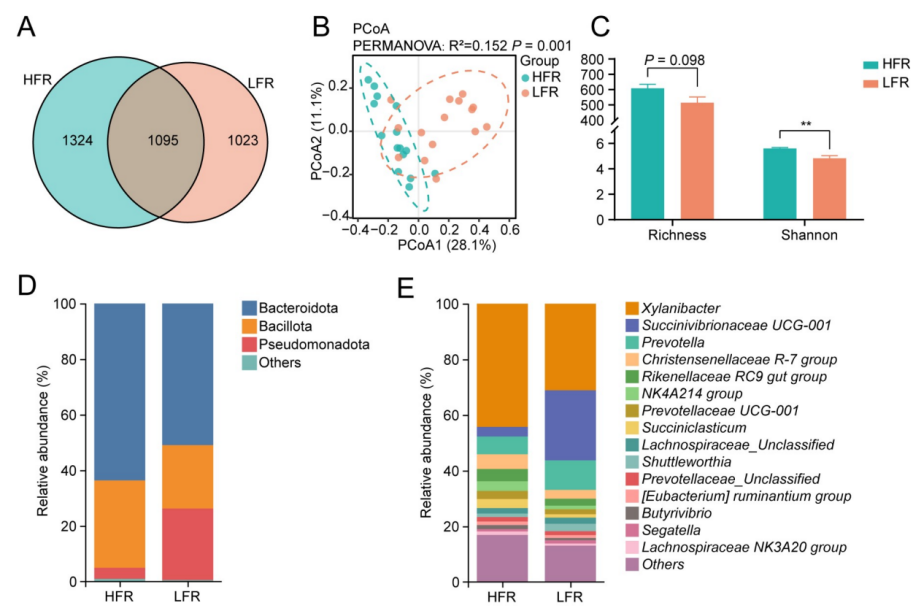
图 扩增子测序揭示两个表型组之间不同的微生物群落结构
文章4 Microbiome (IF=12.7)
厦门市室内空气与健康重点实验室,区域与城市生态国家重点实验室,中国科学院城市环境研究所

研究背景
人们对室内微生物的健康风险认知不足,现有研究多基于相对丰度,缺乏能准确评估暴露风险的绝对定量数据。此外,微生物分泌的毒力因子及其新型载体胞外囊泡在室内环境中的健康影响尚不明确。
实验设计
对全中国118个家庭的灰尘进行采样,并采用多组学方法进行分析:包括(1)16s/ITS绝对定量测序(spike in法);(2)宏基因组学分析毒力因子和抗性基因;(3)宏蛋白质组学验证胞外囊泡携带的毒力蛋白;(4)结合健康问卷与环境数据进行关联分析。
主要研究结果
**1、定量风险:**家庭灰尘中潜在病原菌和真菌的绝对丰度可观,且其绝对丰度与居民鼻炎、喘息、皮炎的患病率正相关。
**2、城乡差异:**城市家庭灰尘中潜在病原菌的相对丰度显著高于农村,但驱动因素是纬度、湿度、温度等环境变量。
**3、新机制:**胞外囊泡是重要的毒力载体,携带了灰尘中近半数的细菌毒力因子和抗性基因,这为理解非活菌的持续健康风险提供了新机制。
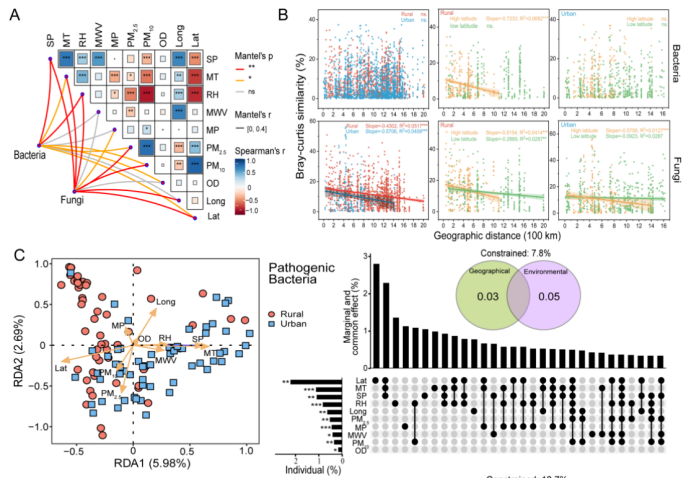
图 家庭灰尘中成形因素的识别
文章5 Nature Communications(IF=15.7)
吉林农业大学动物医学院

研究背景
鸡球虫病由柔嫩艾美耳球虫引起,对全球家禽业构成严重威胁。盲肠是E. tenella的专性寄生部位,其微生物群在宿主-寄生虫相互作用中扮演关键角色。然而,微生物稳态如何具体影响寄生虫发育和宿主抵抗力,其因果机制尚不明确。
实验设计
**核心模型:**使用抗生素诱导的盲肠菌群失调模型,建立伪无菌鸡感染模型。
**验证方法:**通过粪便微生物群移植验证微生物群与寄生虫发育的因果关系。
**关键干预:**鉴定出关键菌属Intestinimonas后,进一步移植其特定菌株I. butyriciproducens,评估其对感染的影响。
**多组学分析:**结合16S rRNA测序、转录组学和代谢组学,分析微生物群落变化、寄生虫基因表达及代谢物差异。
**免疫评估:**通过流式细胞术、ELISA等方法,分析CD8+ T细胞等免疫指标。
主要研究结果
**1、微生物群影响发育:**抗生素诱导的菌群失调显著损害了E. tenella的大配子发生,证明寄生虫发育依赖于微生物群。
**2、关键菌属发现:**Intestinimonas被鉴定为抑制寄生虫发育的关键菌属。其丰度与寄生虫发育呈负相关。
**3、分子机制:**Intestinimonas通过下调寄生虫的EtGFAT基因表达来抑制其大配子形成。EtGFAT是介导大配子形成的关键酶。
**4、功能验证:**移植I. butyriciproducens可减轻感染引起的临床症状(如病变评分、卵囊排出量),并提高抗球虫指数。
**5、免疫增强:**I. butyriciproducens能促进盲肠扁桃体中CD8+ T细胞分泌IFN-γ,从而增强宿主对E. tenella的适应性免疫应答。
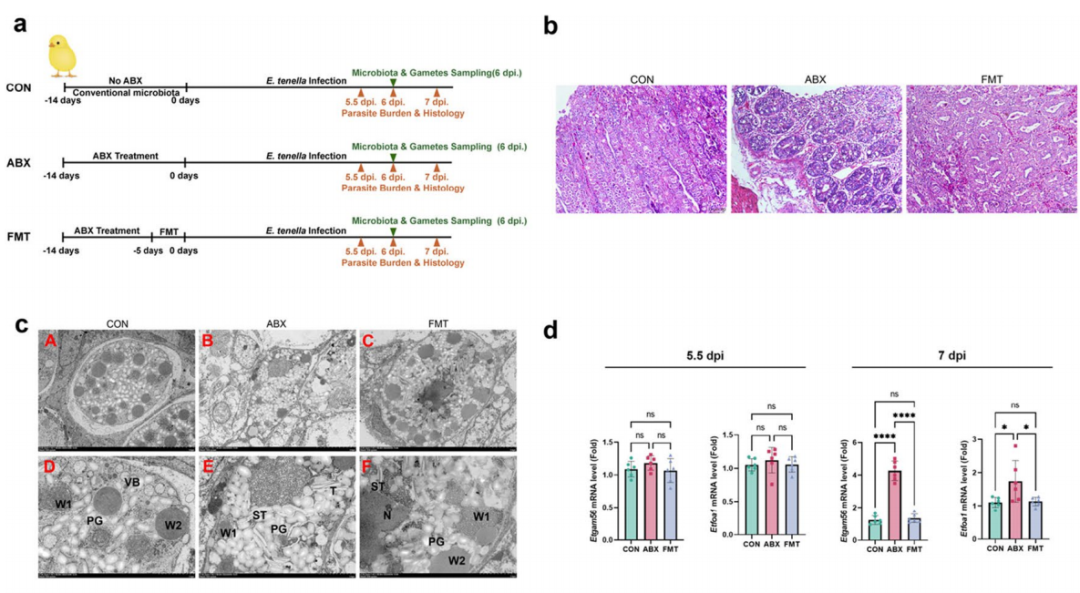
图 FMT 可消除埃坦拉菌引起的发育停滞现象
文章6 Gut(IF=26.2)
中山大学肿瘤防治中心

研究背景
鼻咽癌(NPC)与EB病毒(EBV)感染高度相关,但仅少数EBV感染者会发展为NPC,提示存在其他关键影响因素。肠道微生物组在调节宿主免疫和病毒感染结局中起重要作用,但其与EBV感染及NPC发生发展的潜在联系尚不明确。同时,临床上迫切需要更有效的NPC早期诊断工具。
实验设计
前瞻性纳入516名EBV相关NPC患者和263名健康对照。通过宏基因组测序,分析比较患者与对照的肠道微生物组多样性、物种组成和功能通路差异。通过生存分析,鉴定与NPC患者总生存期相关的预后微生物特征,并探索其与肿瘤免疫微环境(TME)的潜在联系。
主要研究结果
**1、微生物组特征:**NPC患者肠道微生物组显著失调,表现为产短链脂肪酸(SCFA)的菌种(如Roseburia spp., Anaerostipes hadrus)耗竭,以及丁酸盐代谢功能降低,这些变化与更高的血浆EBV DNA载量显著相关。
**2、诊断性能:**基于26个物种标志物构建的随机森林分类器,区分NPC与对照的曲线下面积(AUC)达0.917。当与EBV特异性标志物结合后,诊断性能进一步提升至AUC 0.984,尤其在早期(I-II期)NPC中表现优异(AUC=0.973)。
**3、预后价值:**研究鉴定出16个与预后相关的微生物物种,并构建了预后微生物评分(PMS)。高PMS与更差的总生存期显著相关,并且是独立的预后因素。结合PMS与EBV DNA能更有效地对患者进行风险分层。
**4、与肿瘤微环境关联:**基于PMS和EBV DNA划分的高风险患者,其肿瘤组织表现出更强的免疫抑制性微环境特征。
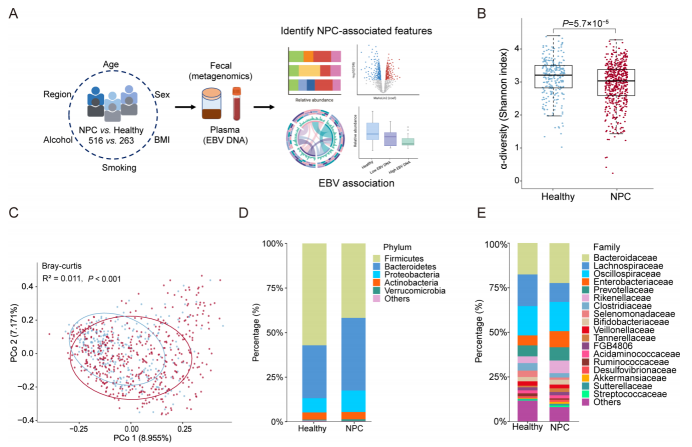
图 研究设计及宏基因组分析结果
文章7 Microbiome(IF=12.7)
南京农业大学资源与环境科学学院

研究背景
当归(Angelica sinensis)是一种重要的药用植物,但其种植常受根腐病(由尖孢镰刀菌 Fusarium oxysporum引起)的严重威胁。植物根际微生物组在植物抗病中起关键作用,植物分泌的次生代谢物是调控根际微生物群落组装的关键信号。然而,代谢组如何驱动微生物组组装并具体决定植物对土传病害的抗性,其机制尚不清楚。
实验设计
**样本对比:**采集田间健康和患病当归的根际土壤,比较其微生物群落和代谢物组成。
**多组学分析:**结合16S rRNA/ITS测序、宏基因组测序和非靶代谢组学,分析微生物组成、功能基因及代谢物差异。
**菌株分离与功能验证:**从土壤中分离镰刀菌和链霉菌菌株,通过盆栽实验验证病原性,并通过体外拮抗实验筛选有益链霉菌。
**机制探究:**通过PCR检测链霉菌的类固醇降解基因;通过转录组学分析脂质信号分子(茉莉酸甲酯、油菜素内酯)对关键链霉菌菌株基因表达的影响。
**干预验证:**在盆栽和田间试验中,评估外源添加合生元(脂质益生元 + 链霉菌益生菌)对防治根腐病的效果。
主要研究结果
**1、微生物组差异:**患病土壤中镰刀菌相对丰度显著升高,而有益菌链霉菌的相对丰度降低。
**2、功能基因富集:**宏基因组分析显示,健康土壤中脂质代谢相关基因(特别是类固醇降解途径基因)显著富集,这些基因主要来源于链霉菌科。
**3、关键代谢物鉴定:**代谢组学发现,健康土壤中脂质类化合物(如茉莉酸甲酯和油菜素内酯)含量更高,且这些代谢物的丰度与链霉菌的丰度呈显著正相关。
**4、有益菌功能:**分离出的链霉菌菌株 S15 (Streptomyces avidinii) 能有效拮抗尖孢镰刀菌,并增强当归根系抗性。S15 基因组中含有类固醇降解基因,并能以茉莉酸甲酯和油菜素内酯为碳源促进生长。
**5、分子机制:**转录组分析表明,茉莉酸甲酯和油菜素内酯能上调 S15 菌株中与链霉素生物合成、生物膜形成和孢子形成相关的基因表达,增强其定殖和抗菌能力。
**6、合生元效果:**外源添加合生元(脂质益生元 + S15)能有效富集根际中与 S15 同源的链霉菌,构建有益的细菌群落,从而减轻镰刀菌胁迫,防治根腐病。
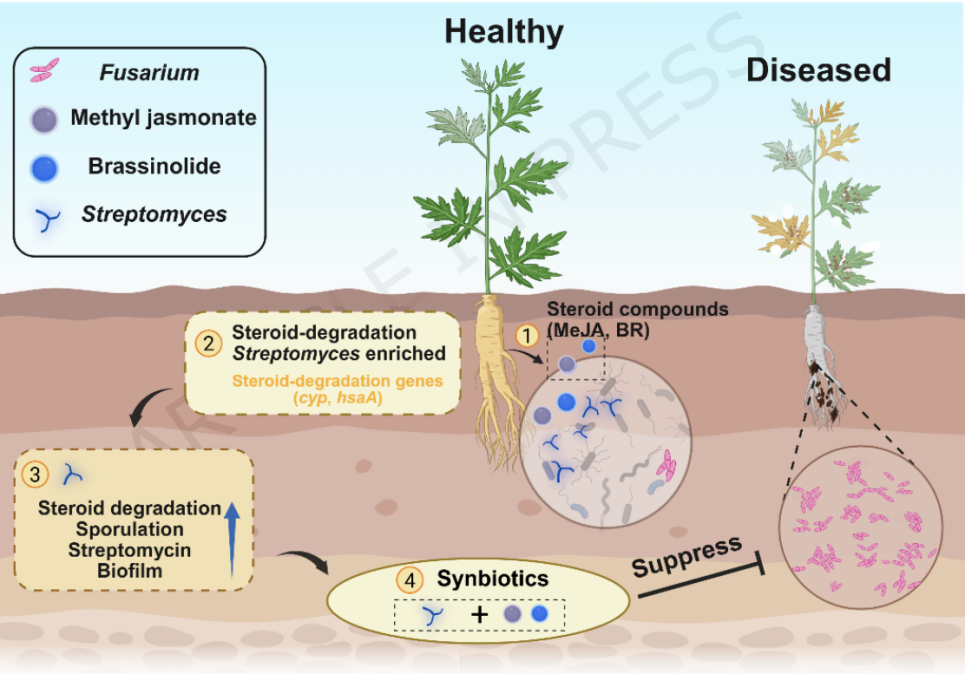
图 甾体诱导的链霉菌如何增强当归根腐病抗性的概念模型
文章8 npj Biofilms and Microbiomes(IF=9.2)
第四军医大学公共卫生学院,教育部特殊作业环境危害评估与防护重点实验室,陕西省环境危害评估与防护重点实验室

研究背景
抗生素耐药性(AMR)是本世纪最紧迫的全球健康挑战之一。抗生素耐药基因(ARGs)作为关键的环境污染物,在复杂的生态界面(野生动物、家畜、媒介和环境)间传播的生态网络仍不清楚。蜱作为专性吸血体外寄生虫,连接着野生动物、家畜和人类,其微生物群可能是ARGs的储存库和传播媒介。本研究基于"One Health"(一体化健康)框架,旨在阐明在青藏高原边缘的生态交错带中,ARGs在野生动物(喜马拉雅旱獭)、媒介(蜱)、家畜(绵羊)及其共享环境(洞穴土壤)之间的传播模式和机制。
实验设计
在祁连山国家级自然保护区(青藏高原边缘的重要生态交错带)进行实地调查。采集了同一生态栖息地内的家养绵羊粪便、蜱及其洞穴土壤样本。进行鸟枪法宏基因组测序以及宏基因组binning。
主要研究结果
1、微生物组组成存在宿主特异性差异:哺乳动物(旱獭和绵羊)肠道微生物组以厚壁菌门(Firmicutes)为主,而蜱和土壤样本则表现出独特的聚类,以变形菌门(Proteobacteria)占主导地位。
2、耐药组机制呈现生态位特异性模式:目标修饰(target alteration)在哺乳动物样本中占主导(旱獭51.9%,绵羊37.7%);蜱则表现出更高的抗生素失活(32.0%)和外排泵机制(31.4%);土壤中以外排泵机制为主(44.2%)。
3、揭示了跨宿主共享的耐药组和功能基因库:蜱与旱獭在物种(71.9%)、ARGs(51.7%)和毒素基因(90.1%)上相似度最高。土壤与哺乳动物宿主(旱獭和绵羊)无共享物种,但分别保留了32.1%的ARGs和超过86%的毒素基因,表明其作为非生命储存库的功能。
来源追踪分析显示,蜱对旱獭和绵羊耐药组的贡献显著(旱獭:0.46-0.63;绵羊:0.29-0.56)。
4、构建了水平基因转移(HGT)网络并评估了ARGs风险。绵羊携带的ARGs风险最高(包含Rank I/II级高风险基因),与拟杆菌科(Bacteroidaceae)等特定细菌谱系相关。
网络分析表明,旱獭和绵羊来源的MAGs是ARGs传播的核心枢纽,而蜱来源的MAGs则可能充当了环境源向动物宿主传播的潜在途径。
5、发现了大量未培养的微生物"暗物质":在鉴定的834个物种级基因组箱(SGBs)中,有263个(31.5%)是未知的SGBs,主要来自土壤和蜱样本,凸显了该生态界面中巨大的未探索微生物多样性。
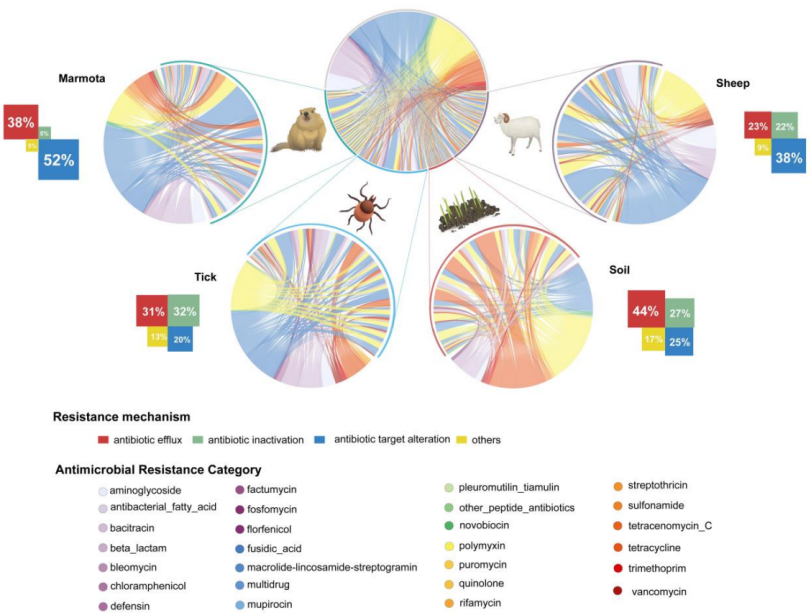
图 MAG的系统发育与分类