引言
在本文中演示了如何合并包含单细胞染色质数据的多个 Seurat 对象。为了进行演示,将使用 10x Genomics 提供的四个 scATAC-seq PBMC 数据集:
实战
在整合多个单细胞染色质数据集的过程中,应当意识到,如果对每个数据集单独进行了峰值检测,那么检测到的峰值可能并不完全一致。为此,需要构建一个适用于所有合并数据集的共通峰值集合。
可以通过GenomicRanges包提供的功能来实现这一点。该包中的reduce函数能够将所有相互重叠的峰值进行合并。此外,disjoin函数也是一个不错的选择,它能够生成互不重叠的独立峰值集合。以下是一个图解示例,用以展示reduce和disjoin两种方法的差异:
gr <- GRanges(seqnames = "chr1", ranges = IRanges(start = c(20, 70, 300), end = c(120, 200, 400)))
gr
## GRanges object with 3 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 20-120 *
## [2] chr1 70-200 *
## [3] chr1 300-400 *
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths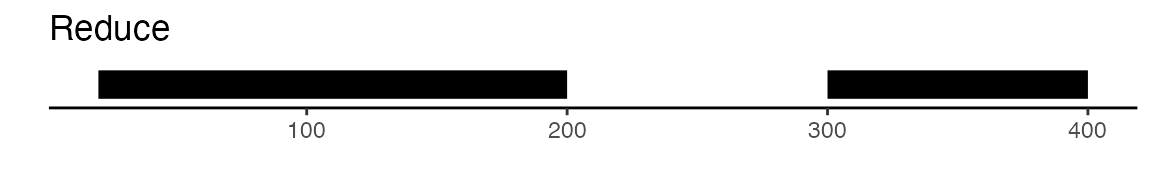

构建统一的峰值集合
如果在各个实验中分别鉴定了峰值,那么这些峰值可能并不完全一致。可以通过整合所有实验数据中的峰值,来形成一个统一的峰值集合,并在合并这些数据集之前,对每个实验中的峰值集合进行量化分析。
首先,需要导入每个实验的峰值位置信息,并将其转换成基因组范围内的格式。接着,利用 GenomicRanges 包中的 reduce 函数,来创建一个适用于所有数据集的峰值集合,以便在每个实验中进行量化分析。
library(Signac)
library(Seurat)
library(GenomicRanges)
library(future)
plan("multicore", workers = 4)
options(future.globals.maxSize = 50000 * 1024^2) # for 50 Gb RAM
# read in peak sets
peaks.500 <- read.table(
file = "pbmc500/atac_pbmc_500_nextgem_peaks.bed",
col.names = c("chr", "start", "end")
)
peaks.1k <- read.table(
file = "pbmc1k/atac_pbmc_1k_nextgem_peaks.bed",
col.names = c("chr", "start", "end")
)
peaks.5k <- read.table(
file = "pbmc5k/atac_pbmc_5k_nextgem_peaks.bed",
col.names = c("chr", "start", "end")
)
peaks.10k <- read.table(
file = "pbmc10k/atac_pbmc_10k_nextgem_peaks.bed",
col.names = c("chr", "start", "end")
)
# convert to genomic ranges
gr.500 <- makeGRangesFromDataFrame(peaks.500)
gr.1k <- makeGRangesFromDataFrame(peaks.1k)
gr.5k <- makeGRangesFromDataFrame(peaks.5k)
gr.10k <- makeGRangesFromDataFrame(peaks.10k)
# Create a unified set of peaks to quantify in each dataset
combined.peaks <- reduce(x = c(gr.500, gr.1k, gr.5k, gr.10k))
# Filter out bad peaks based on length
peakwidths <- width(combined.peaks)
combined.peaks <- combined.peaks[peakwidths < 10000 & peakwidths > 20]
combined.peaks
## GRanges object with 89951 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 565153-565499 *
## [2] chr1 569185-569620 *
## [3] chr1 713551-714783 *
## [4] chr1 752418-753020 *
## [5] chr1 762249-763345 *
## ... ... ... ...
## [89947] chrY 23422151-23422632 *
## [89948] chrY 23583994-23584463 *
## [89949] chrY 23602466-23602779 *
## [89950] chrY 28816593-28817710 *
## [89951] chrY 58855911-58856251 *
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengths构建片段对象
为了对合并的峰值集合进行量化分析,需要针对每个实验创建一个片段对象。片段对象是一个在 Signac 中特别定义的类,它负责存储与单个片段文件相关的所有数据。
首先需要导入每个实验的细胞元数据,这样就能了解每个文件包含哪些细胞的条形码信息。之后,利用 CreateFragmentObject 函数来生成片段对象。这个函数会进行一系列验证,确保文件不仅存在于硬盘上,而且已经过压缩和索引处理,同时计算文件及其 tabix 索引的 MD5 校验值,以便于能够检测到文件在任何时间点的变更情况,并确认文件中确实包含了预期的细胞类型。
# load metadata
md.500 <- read.table(
file = "pbmc500/atac_pbmc_500_nextgem_singlecell.csv",
stringsAsFactors = FALSE,
sep = ",",
header = TRUE,
row.names = 1
)[-1, ] # remove the first row
md.1k <- read.table(
file = "pbmc1k/atac_pbmc_1k_nextgem_singlecell.csv",
stringsAsFactors = FALSE,
sep = ",",
header = TRUE,
row.names = 1
)[-1, ]
md.5k <- read.table(
file = "pbmc5k/atac_pbmc_5k_nextgem_singlecell.csv",
stringsAsFactors = FALSE,
sep = ",",
header = TRUE,
row.names = 1
)[-1, ]
md.10k <- read.table(
file = "pbmc10k/atac_pbmc_10k_nextgem_singlecell.csv",
stringsAsFactors = FALSE,
sep = ",",
header = TRUE,
row.names = 1
)[-1, ]
# perform an initial filtering of low count cells
md.500 <- md.500[md.500$passed_filters > 500, ]
md.1k <- md.1k[md.1k$passed_filters > 500, ]
md.5k <- md.5k[md.5k$passed_filters > 500, ]
md.10k <- md.10k[md.10k$passed_filters > 1000, ] # sequenced deeper so set higher cutoff
# create fragment objects
frags.500 <- CreateFragmentObject(
path = "pbmc500/atac_pbmc_500_nextgem_fragments.tsv.gz",
cells = rownames(md.500)
)
frags.1k <- CreateFragmentObject(
path = "pbmc1k/atac_pbmc_1k_nextgem_fragments.tsv.gz",
cells = rownames(md.1k)
)
frags.5k <- CreateFragmentObject(
path = "pbmc5k/atac_pbmc_5k_nextgem_fragments.tsv.gz",
cells = rownames(md.5k)
)
frags.10k <- CreateFragmentObject(
path = "pbmc10k/atac_pbmc_10k_nextgem_fragments.tsv.gz",
cells = rownames(md.10k)
)在各数据集中对峰值进行量化
利用 FeatureMatrix 函数,现在能够为每个样本生成一个以峰值和细胞为维度的矩阵。此函数通过 future 包支持并行计算。
pbmc500.counts <- FeatureMatrix(
fragments = frags.500,
features = combined.peaks,
cells = rownames(md.500)
)
pbmc1k.counts <- FeatureMatrix(
fragments = frags.1k,
features = combined.peaks,
cells = rownames(md.1k)
)
pbmc5k.counts <- FeatureMatrix(
fragments = frags.5k,
features = combined.peaks,
cells = rownames(md.5k)
)
pbmc10k.counts <- FeatureMatrix(
fragments = frags.10k,
features = combined.peaks,
cells = rownames(md.10k)
)总结
本文[1]提供了一个详细的流程来合并单细胞染色质数据集,包括数据下载、预处理、合并以及后续的分析和可视化步骤。强调了在合并过程中创建共有峰值集合的重要性,并提供了在没有片段文件时的替代方法。
Reference 1
Source: https://stuartlab.org/signac/articles/merging
本文由mdnice多平台发布