关注 M r . m a t e r i a l , \color{Violet} \rm Mr.material\ , Mr.material , 更 \color{red}{更} 更 多 \color{blue}{多} 多 精 \color{orange}{精} 精 彩 \color{green}{彩} 彩!
主要专栏内容包括:
†《LAMMPS小技巧》: ‾ \textbf{ \underline{\dag《LAMMPS小技巧》:}} †《LAMMPS小技巧》: 主要介绍采用分子动力学( L a m m p s Lammps Lammps)模拟相关安装教程、原理以及模拟小技巧(难度: ★ \bigstar ★)
††《LAMMPS实例教程---In文件详解》: ‾ \textbf{ \underline{\dag\dag《LAMMPS实例教程---In文件详解》:}} ††《LAMMPS实例教程---In文件详解》: 主要介绍采用分子动力学( L a m m p s Lammps Lammps)模拟相关物理过程模拟。(包含:热导率计算、定压比热容计算,难度: ★ \bigstar ★ ★ \bigstar ★ ★ \bigstar ★)
†††《Lammps编程技巧及后处理程序技巧》: ‾ \textbf{ \underline{\dag\dag\dag《Lammps编程技巧及后处理程序技巧》:}} †††《Lammps编程技巧及后处理程序技巧》: 主要介绍针对分子模拟的动力学过程(轨迹文件)进行后相关的处理分析(需要一定编程能力。难度: ★ \bigstar ★ ★ \bigstar ★ ★ \bigstar ★ ★ \bigstar ★ ★ \bigstar ★)。
††††《分子动力学后处理集成函数---Matlab》: ‾ \textbf{ \underline{\dag\dag\dag\dag《分子动力学后处理集成函数---Matlab》:}} ††††《分子动力学后处理集成函数---Matlab》: 主要介绍针对后处理过程中指定函数,进行包装,方便使用者直接调用(需要一定编程能力,难度: ★ \bigstar ★ ★ \bigstar ★ ★ \bigstar ★ ★ \bigstar ★)。
†††††《SCI论文绘图---Python绘图常用模板及技巧》: ‾ \textbf{ \underline{\dag\dag\dag\dag\dag《SCI论文绘图---Python绘图常用模板及技巧》:}} †††††《SCI论文绘图---Python绘图常用模板及技巧》: 主要介绍针对处理后的数据可视化,并提供对应的绘图模板(需要一定编程能力,难度: ★ \bigstar ★ ★ \bigstar ★ ★ \bigstar ★ ★ \bigstar ★)。
††††††《分子模拟---Ovito渲染案例教程》: ‾ \textbf{ \underline{\dag\dag\dag\dag\dag\dag《分子模拟---Ovito渲染案例教程》:}} ††††††《分子模拟---Ovito渲染案例教程》: 主要采用 O v i t o \rm Ovito Ovito软件,对 L a m m p s \rm Lammps Lammps 生成的轨迹文件进行渲染(难度: ★ \bigstar ★ ★ \bigstar ★)。
专栏说明(订阅后可浏览对应专栏全部博文): ‾ \color{red}{\textbf{ \underline{专栏说明(订阅后可浏览对应专栏全部博文):}}} 专栏说明(订阅后可浏览对应专栏全部博文):
注意: \color{red} 注意: 注意:如需只订阅某个单独博文,请联系博主邮箱咨询。 l a m m p s _ m a t e r i a l s @ 163. c o m \rm lammps\_materials@163.com lammps_materials@163.com
♠ \spadesuit ♠ † \dag † 开源后处理集成程序:请关注专栏《LAMMPS后处理------MATLAB子函数合集整理》
♠ \spadesuit ♠ † \dag † † \dag † 需要付费定制后处理程序请邮件联系: l a m m p s _ m a t e r i a l s @ 163. c o m \rm lammps\_materials@163.com lammps_materials@163.com
建模篇:分子动力学模型快速构建 \rm 建模篇:分子动力学模型快速构建 建模篇:分子动力学模型快速构建
M a t e r i a l S t u d i o + M o l t e m p l a t e + P a c k m o l \rm \color{red} Material\ Studio+Moltemplate+Packmol Material Studio+Moltemplate+Packmol
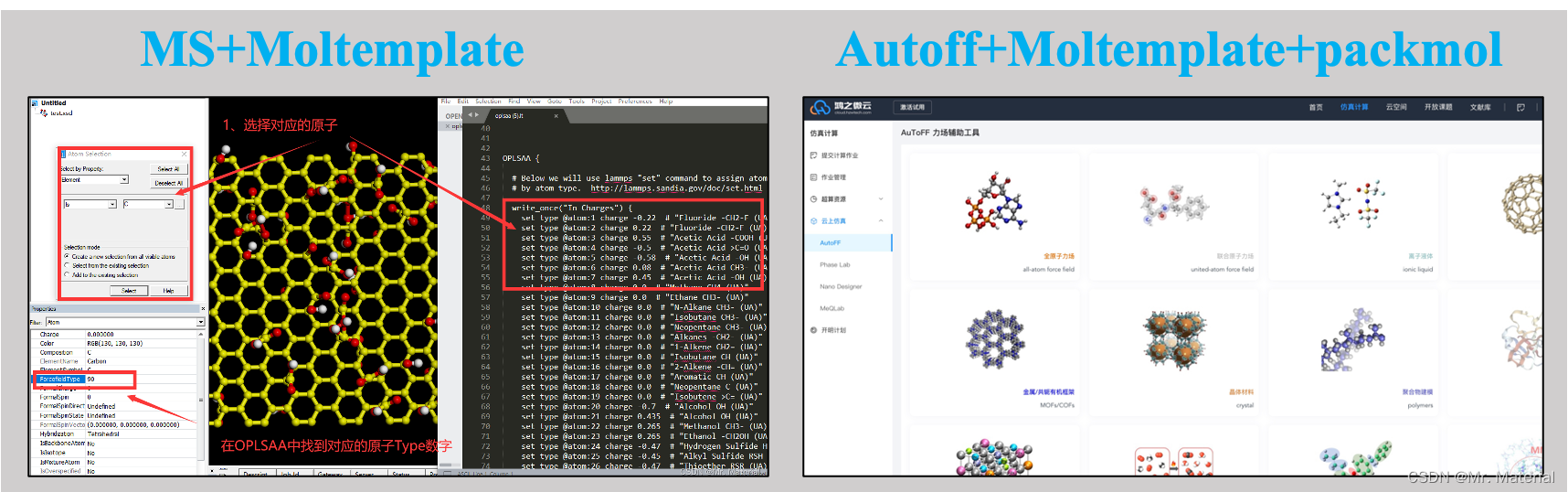
1、 建模篇(上):OPLSAA力场参数之快速建模---MS+Moltemplate
2 、建模篇(下):力场快速设置-AuToFF+Moltemplate
反应力场的生成物、反应路径分析方法
- [建模篇:分子动力学模型快速构建 \rm 建模篇:分子动力学模型快速构建 建模篇:分子动力学模型快速构建](#建模篇:分子动力学模型快速构建 \rm 建模篇:分子动力学模型快速构建 建模篇:分子动力学模型快速构建)
-
-
- [1、 建模篇(上):OPLSAA力场参数之快速建模---MS+Moltemplate(https://blog.csdn.net/qq_43689832/article/details/127913687)](#1、 建模篇(上):OPLSAA力场参数之快速建模—MS+Moltemplate)
- [2 、建模篇(下):力场快速设置-AuToFF+Moltemplate(https://blog.csdn.net/qq_43689832/article/details/137957981)](#2 、建模篇(下):力场快速设置-AuToFF+Moltemplate)
-
- [一、LAMMPS in文件(出自Example/reax/CHO)](#一、LAMMPS in文件(出自Example/reax/CHO))
- 二、化学键生成可视化
-
- [1. Python脚本](#1. Python脚本)
- [2. Matlab 脚本](#2. Matlab 脚本)
- 三、反应路径分析
-
- [1. 安装](#1. 安装)
- [2. 分析](#2. 分析)
-
- [2.1 反应网络](#2.1 反应网络)
- [2.2 生成物](#2.2 生成物)
- [2.3 路径](#2.3 路径)
- [四 详细案例及教程](#四 详细案例及教程)
针对 《 M a t l a b 实现反应力场物产统计》 \rm 《Matlab实现反应力场物产统计》 《Matlab实现反应力场物产统计》 中的案例( E x a m p l e / r e a x / C H O \rm Example/reax/CHO Example/reax/CHO)。
一、LAMMPS in文件(出自Example/reax/CHO)
根据Lammps以下的命令可以获得化学键的断裂情况以及反应生成物,那么如何统计反应路径呢?

cpp
# REAX potential for CHO system
# .....
units real
atom_style charge
read_data data.CHO
pair_style reax/c NULL
pair_coeff * * ffield.reax.cho H C O
neighbor 2 bin
neigh_modify every 10 delay 0 check no
fix 1 all nve
fix 2 all qeq/reax 1 0.0 10.0 1e-6 param.qeq
fix 3 all temp/berendsen 500.0 5000.0 100.0
fix 4 all reax/c/species 1 1 100 species.out
timestep 0.25
dump 1 all atom 1000 dump.reax.cho
run 100000二、化学键生成可视化

1. Python脚本
这里用到了鲍老师(公众号: LAMMPS与AI材料设计)早起开源的代码计算,结合Ovito可以实现化学键断裂、生成的可视化。

python
#结合LAMMPS中fix bond/reaxff和dump命令的输出文件,将fix bond/reaxff中的键结信息和dump中的坐标信息转换组合写入一系列data文件中。由此就可利用ovito进行可视化。ovito可以读入系列文件,这样就可以看到分子如何断键分解,或反应物如何成键生成产物
#要求使用atom_style full,以保留分子id信息在ovito中分析
#fix bond/reaxff和dump的输出频率要一致。当模拟时间很长时输出频率不能太快。
#对于原子数很多的体系代码速度会较慢
#dump命令必须使用以下格式
#dump 1 all custom 1000 dump.lammpstrj id mol type q x y z
#
work_path = "F:\\官能团\\out-12-16\\group-120-150\\新建文件夹\\New Folder\\"
bonds = open(work_path + "bonds.reaxff","r")
dump = open(work_path + "dump.out","r")
N_Frame = 49
n_atom = 105
n_bond = 0
atom_sec = []
bond_sec = []
for i in range(N_Frame):
for j in range(5):
line_dump = dump.readline()
line_dump = dump.readline()
xlo = line_dump.split()[0]
xhi = line_dump.split()[1]
line_dump = dump.readline()
ylo = line_dump.split()[0]
yhi = line_dump.split()[1]
line_dump = dump.readline()
zlo = line_dump.split()[0]
zhi = line_dump.split()[1]
line_dump = dump.readline()
for k in range(n_atom):
line_dump = dump.readline()
atom_sec.append(line_dump)
if i == 0:
pre_line = 7
else:
pre_line = 8
for j in range(pre_line):
line_bond = bonds.readline()
for k in range(n_atom):
line_bond = bonds.readline()
nb = int(line_bond.split()[2])
for l in range(nb):
n_bond += 1
tmp = str(n_bond) + " " + "1" + " " + line_bond.split()[0] + " " + line_bond.split()[3+l] + "\n"
bond_sec.append(tmp)
data = open(work_path + "data" + str(i),"w")
data.write("LAMMPS data from bonds\n")
data.write("\n")
data.write(str(n_atom) + " " + "atoms\n")
data.write("3 atom types\n")
data.write(str(n_bond) + " " + "bonds\n")
data.write("1 bond types\n")
data.write("\n")
data.write(str(xlo) + " " + str(xhi) + " " + "xlo xhi\n")
data.write(str(ylo) + " " + str(yhi) + " " + "ylo yhi\n")
data.write(str(zlo) + " " + str(zhi) + " " + "zlo zhi\n")
data.write("\n")
data.write("Atoms\n")
data.write("\n")
data.writelines(atom_sec)
data.write("\n")
data.write("Bonds\n")
data.write("\n")
data.writelines(bond_sec)
data.close()
n_bond = 0
atom_sec = []
bond_sec = []2. Matlab 脚本
matlab
work_path = "F:\官能团\out-12-16\group-120-150\新建文件夹\New Folder\";
% 使用 fullfile 拼接路径
bonds_file_path = fullfile(work_path, "bonds.reaxff");
dump_file_path = fullfile(work_path, "dump.out");
% 检查文件是否存在
if ~isfile(bonds_file_path)
error("File not found: %s", bonds_file_path);
end
if ~isfile(dump_file_path)
error("File not found: %s", dump_file_path);
end
% 打开文件
bonds_file = fopen(bonds_file_path, 'r');
dump_file = fopen(dump_file_path, 'r');
N_Frame = 49; % 总帧数
n_atom = 105; % 原子数
% 初始化
atom_sec = {};
bond_sec = {};
n_bond = 0;
for i = 1:N_Frame
% 跳过前5行
for j = 1:5
fgetl(dump_file);
end
% 读取盒子尺寸
line_dump = fgetl(dump_file);
line_dump_split = strsplit(line_dump);
xlo = str2double(line_dump_split{1});
xhi = str2double(line_dump_split{2});
line_dump = fgetl(dump_file);
line_dump_split = strsplit(line_dump);
ylo = str2double(line_dump_split{1});
yhi = str2double(line_dump_split{2});
line_dump = fgetl(dump_file);
line_dump_split = strsplit(line_dump);
zlo = str2double(line_dump_split{1});
zhi = str2double(line_dump_split{2});
fgetl(dump_file); % 跳过一行
% 读取原子部分
for k = 1:n_atom
atom_sec{k} = fgetl(dump_file);
end
% bonds.reaxff 文件处理
if i == 1
pre_line = 7;
else
pre_line = 8;
end
for j = 1:pre_line
fgetl(bonds_file);
end
% 读取键信息
bond_idx = 1;
for k = 1:n_atom
line_bond = fgetl(bonds_file);
bond_data = strsplit(line_bond);
nb = str2double(bond_data{3});
for l = 1:nb
n_bond = n_bond + 1;
bond_sec{bond_idx} = sprintf('%d 1 %s %s\n', n_bond, bond_data{1}, bond_data{3+l});
bond_idx = bond_idx + 1;
end
end
% 写入 data 文件
data_filename = fullfile(work_path, sprintf("data%d", i));
data_file = fopen(data_filename, 'w');
if data_file == -1
error("Failed to open file: %s", data_filename);
end
fprintf(data_file, "LAMMPS data from bonds\n\n");
fprintf(data_file, "%d atoms\n", n_atom);
fprintf(data_file, "3 atom types\n");
fprintf(data_file, "%d bonds\n", n_bond);
fprintf(data_file, "1 bond types\n\n");
fprintf(data_file, "%f %f xlo xhi\n", xlo, xhi);
fprintf(data_file, "%f %f ylo yhi\n", ylo, yhi);
fprintf(data_file, "%f %f zlo zhi\n\n", zlo, zhi);
fprintf(data_file, "Atoms\n\n");
fprintf(data_file, "%s\n", atom_sec{:});
fprintf(data_file, "\nBonds\n\n");
fprintf(data_file, "%s\n", bond_sec{:});
fclose(data_file);
% 重置变量
n_bond = 0;
atom_sec = {};
bond_sec = {};
end
% 关闭文件
fclose(bonds_file);
fclose(dump_file);三、反应路径分析
这里需要用到,曾晋哲等开发的《Reaxff力场反应网络分析reacnetgenerator》。
Jinzhe Zeng, Liqun Cao, Chih-Hao Chin, Haisheng Ren, John Z. H. Zhang, and Tong Zhu. ReacNetGenerator: an automatic reaction network generator for reactive molecular dynamics simulations. Phys. Chem. Chem. Phys., 22(2):683--691, 2020. doi:10.1039/C9CP05091D.
1. 安装
cpp
# upgrade pip as old pip may not be supported
pip install -U pip
pip install reacnetgenerator

2. 分析
注意 : \color{red} 注意: 注意: \\rm 轨迹文件格式: id type x y z
针对Lammps生成的轨迹文件
cpp
reacnetgenerator --type bond -i bonds.reaxc -a C H O --nohmm
会在目录下生成一下文件:

2.1 反应网络

2.2 生成物

2.3 路径

四 详细案例及教程
