同学们,大家好!今天给大家介绍一篇研究性论文,原苏木素A通过靶向ACSL4/FTH1轴依赖性铁凋亡保护阿霉素诱导的心肌损伤和心功能障碍,想了解这方面的同学们可以重点关注一下。这篇文章是2024年7月份发表在Advanced Science(IF:14.3,中科院分区:材料科学1区)上的,下面我们详细看看。

在该文章中作者研究了原苏木素A对阿霉素诱导的心肌损伤和心功能障碍的保护作用,并对其作用机理进行了研究,得出以下结论:
1.PrA减轻DOX诱导的心肌损伤和心功能障碍
2.PrA抑制DOX诱导的心脏铁凋亡
3.PrA减轻阿霉素诱导的心肌细胞铁凋亡并维持线粒体功能
4.ACSL4和FTH1被鉴定为直接PrA结合靶点
5.PrA通过抑制ACSL4磷酸化和激活抑制DOX诱导的铁凋亡
6.PrA通过减少溶酶体中FTH1的自噬降解保护DOX介导的铁凋亡
7.PrA介导的铁凋亡抑制减轻缺血再灌注诱导的心脏损伤和功能障碍
作者也在Discussion部分中,对所得出的结论进行逐一分析,并在Experimental Section部分对实验使用药品、小鼠及造模情况进行说明。以下为论文详细内容。
Protosappanin A Protects DOX-Induced Myocardial Injury and Cardiac Dysfunction by Targeting ACSL4/FTH1 Axis-Dependent Ferroptosis
摘要:
阿霉素(DOX)是一种有效的抗癌药物,但其临床应用受到剂量依赖性心脏毒性的限制,部分原因是心肌细胞铁凋亡。然而,开发心脏保护药物以对抗铁缺乏的进展遇到了障碍。原苏木素A(Protosappanin A,PrA)是一种从苏木素中提取的抗炎化合物,具有抗阿霉素诱导的心肌病(DOX-induced cardiomyopathy,DIC)的作用。在这里,据报道,PrA通过减少DOX诱导的铁凋亡和维持线粒体稳态来减轻心肌损伤和功能障碍。随后,通过蛋白质组芯片、分子对接和动力学模拟等方法,确定了PrA的分子靶点。在机制上,PrA与亚铁代谢相关蛋白酰基辅酶A合成酶长链家族成员4(ACSL4)和铁蛋白重链1(FTH1)物理结合,最终抑制ACSL4磷酸化和随后的磷脂过氧化,同时还防止FTH1自噬降解和随后的铁离子(Fe2+)释放。鉴于铁凋亡在缺血再灌注损伤发病机制中的重要作用,本研究进一步推测PrA可通过抑制铁凋亡对IR诱导的心脏损伤产生保护作用。总的来说,一种新的药理学抑制剂被发现,靶向铁凋亡,并揭示了DIC中心肌细胞铁凋亡的双重调节机制,突出了化学药物诱导的心脏毒性和铁凋亡触发的疾病的额外治疗选择。
1. Introduction
蒽环类药物如阿霉素(DOX)在治疗实体瘤和血液恶性肿瘤中是非常有效的。尽管如此,化疗药物的心脏毒性作用严重影响患者的生活质量,表现为心律失常、不可逆的心室功能障碍和长期暴露于药物后的心力衰竭。DOX-induced cardiomysis(DIC)的发生往往预示着预后不良。据报道,在累积剂量为700 mg m-2时,DOX相关心力衰竭的发生率接近25%。虽然DIC的发病机制已经阐明,但目前仍缺乏针对性的预防和治疗策略。
铁死亡是由膜磷脂过氧化引起的一种新型细胞死亡过程。与其他形式的细胞死亡不同,铁死亡主要表现为线粒体疾病、铁代谢异常、脂质过氧化过度和谷胱甘肽过氧化物酶4(GPX4)/谷胱甘肽(GSH)轴失衡。新出现的证据表明,铁死亡与DOX介导的心功能不全密切相关。最近,在DOX攻击的心肌细胞中发现了铁死亡表型。基于上述发现,开发针对铁死亡的靶向药物有可能减轻 DOX 引起的心肌损伤并改善患者的临床结果。
右雷佐生(DXZ)是一种广泛批准的心脏保护剂,用于治疗蒽环类药物介导的心脏毒性,靶向螯合的细胞内亚铁离子,并防止芬顿反应介导的铁凋亡。然而,由于DXZ的副作用,如骨髓抑制和对化疗药物介导的抗肿瘤作用的抵抗,DXZ对癌症生存期的治疗效果存在争议。因此,开发安全有效的治疗策略来对抗化疗药物引起的心脏毒性是非常重要的。原苏木素A(PrA)是从中国传统苏木中提取的一种有效成分,具有抗氧化、抗炎、抗凋亡等多种药理活性。我们前期的研究表明PrA可通过NF-κ B和Akt/mTOR对心脏移植和自身免疫性心肌炎产生免疫抑制作用。尽管最近的进展,PrA在DIC中的作用及其与铁凋亡的关系仍不清楚。
因此,本研究旨在探讨PrA对DIC的影响及其作用机制,并重点探讨其对铁下垂的影响。我们发现PrA可保护DOX诱导的心肌损伤并减轻缺铁症诱导的线粒体功能障碍。在机制上,PrA直接结合酰基辅酶A合成酶长链家族成员4(ACSL4)和铁蛋白重链1(FTH1),阻止ACSL4磷酸化和FTH1自噬降解,从而抑制心肌细胞铁蛋白下垂。由于缺血再灌注(I/R)诱导的心功能障碍可通过抑制铁细胞凋亡而减轻,因此我们进一步证实PrA减轻I/R介导的铁细胞凋亡和心脏损伤。总之,我们的数据为临床治疗化疗药物诱导的心脏毒性和其他心血管相关疾病提供了新的思路。
2. Results
2.1.PrA减轻DOX诱导的心肌损伤和心功能障碍
为了研究PrA在DOX诱导的心脏毒性中的作用,我们建立了一个DICmurine模型,并用PrA(5mg/kg,20mg/kg)或DXZ处理指定的天数(图1A,B)。与DXZ相似,PrA以剂量依赖性方式显著提高了DOX处理小鼠的存活率(图1C)。超声心动图评估显示,与对照组相比,DIC小鼠的心功能显著下降,主要反映在左心室射血分数(LVEF)、左心室短轴缩短率(FS)、左心室舒张期内径(LVIDd)和左心室收缩期内径(LVIDs)参数上。相反,在用DXZ或增加PrA剂量治疗后观察到心脏功能的部分恢复,表明对DOX诱导的心功能障碍具有保护作用(图1D-H)。我们通过分析心肌损伤标志物和心脏组织病理学改变来评估PrA对DOX诱导的心肌损伤的影响。生化分析显示,PrA处理以剂量-反应方式显著抑制DOX诱导的血清肌酸激酶-MB(CK-MB)和乳酸脱氢酶(LDH)水平的升高(图1 I,J)。苏木精和伊红(H&E)染色显示DIC小鼠中有严重的炎性浸润,增加PrA浓度可缓解(图1 K)。此外,通过麦胚凝集素(WGA)、天狼星红和Masson三色染色法测定,PrA剂量依赖性地抑制DOX诱导的小鼠心肌细胞肥大和心肌纤维化(图1 L-Q)。TUNEL染色证实,DOX诱导的心肌DNA损伤通过PrA处理得到改善(图1 R、S)。值得注意的是,20 mg/kg的高浓度PrA在DIC中显示出心脏保护功效,部分超过DXZ,主要反映了对DOX介导的血清CK-MB和LDH水平的更好抑制(图1 I,J)。考虑到DOX常用于乳腺癌的治疗,我们使用雌性小鼠对上述指标进行了复检,获得了与雄性小鼠相似的结果(图S1 A-H,N-P,支持性信息)。综上所述,我们的数据表明PrA减轻了DOX诱导的心脏损伤并防止了心功能障碍。
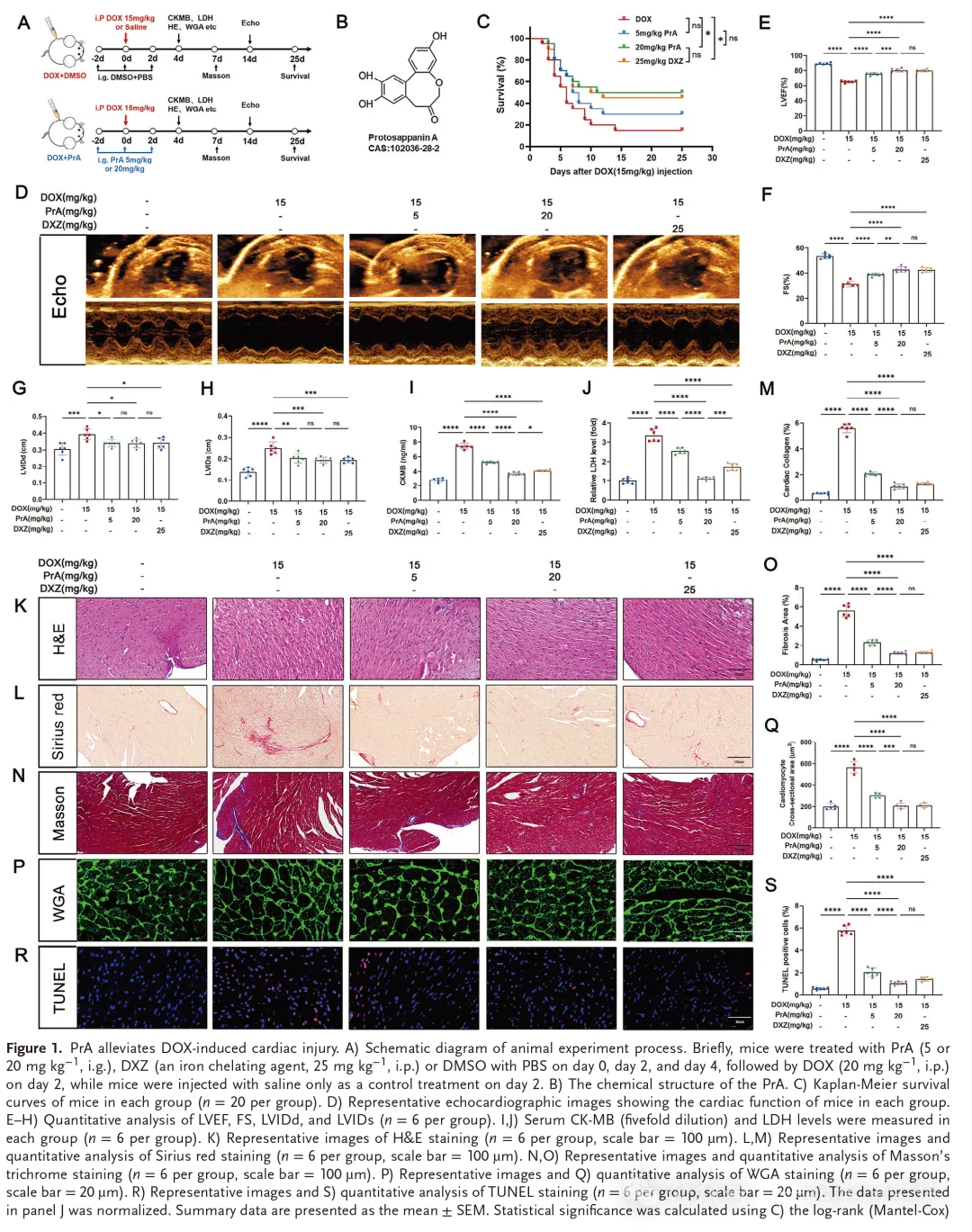
2.2.PrA抑制DOX诱导的心脏铁凋亡
为了揭示PrA对DIC保护作用的分子机制,对三组小鼠心脏进行了RNA测序:对照组、DOX组和DOX联合PrA治疗组(20mg/kg)。指定比较组的差异表达基因(DEG)见图2A和图S2 A、B(支持性信息)。如KEGG分析所揭示的(图2B),与对照组相比,在DOX条件组中,铁凋亡途径是这些DEG中最高的富集,支持铁凋亡和DIC之间的密切关系。相反,PrA处理显著降低了DOX诱导的亚铁粪病相关DEGs富集(图2C和图S2C,支持性信息)。此外,基因组富集分析(GSEA)证实DOX上调铁细胞凋亡,但PrA下调铁细胞凋亡(图2D),暗示PrA可通过抑制铁细胞凋亡来保护免受DIC。
为了验证RNA测序的数据,我们评估了PrA对DIC小鼠铁死亡的影响。使用PCR分析小鼠心脏中铁死亡标志物Ptsg2的表达。如预期的那样,PrA剂量依赖性地逆转DOX诱导的Ptsg 2基因表达(图2E和图S1I)。随后,我们评估了小鼠心脏中与铁蛋白增多症相关的必需调节蛋白的丰度,包括GPX4、ACSL4和FTH1。结果表明,与对照组相比,DIC小鼠心脏组织中ACSL4表达量增加,而FTH1和GPX4表达量减少。PrA给药逆转了小鼠中DOX介导的这些蛋白质水平的改变(图2F和图S2D-F和S1J-M)。免疫荧光进一步支持免疫印迹结果(图2G和图S2G-I)。
鉴于铁超载、脂质过氧化和细胞内活性氧(ROS)积累是铁细胞增多症的关键特征,我们进一步检测了上述条件小鼠体内脂质过氧化产物丙二醛(MDA)和谷胱甘肽(GSH)的水平。与对照组相比,DIC小鼠的GSH心脏水平降低,MDA心脏和血清水平升高。相反,PrA逆转了DOX诱导的MDA和GSH水平变化(图2 H-J)。此外,DOX给药后,心脏组织中的总铁和ROS水平显著增加,PrA治疗消除了这些DOX诱导的结果(图2K-N)。总之,我们的数据提示PrA负性调节DIC小鼠的心脏铁蛋白沉着。

2.3.PrA减轻阿霉素诱导的心肌细胞铁凋亡并维持线粒体功能
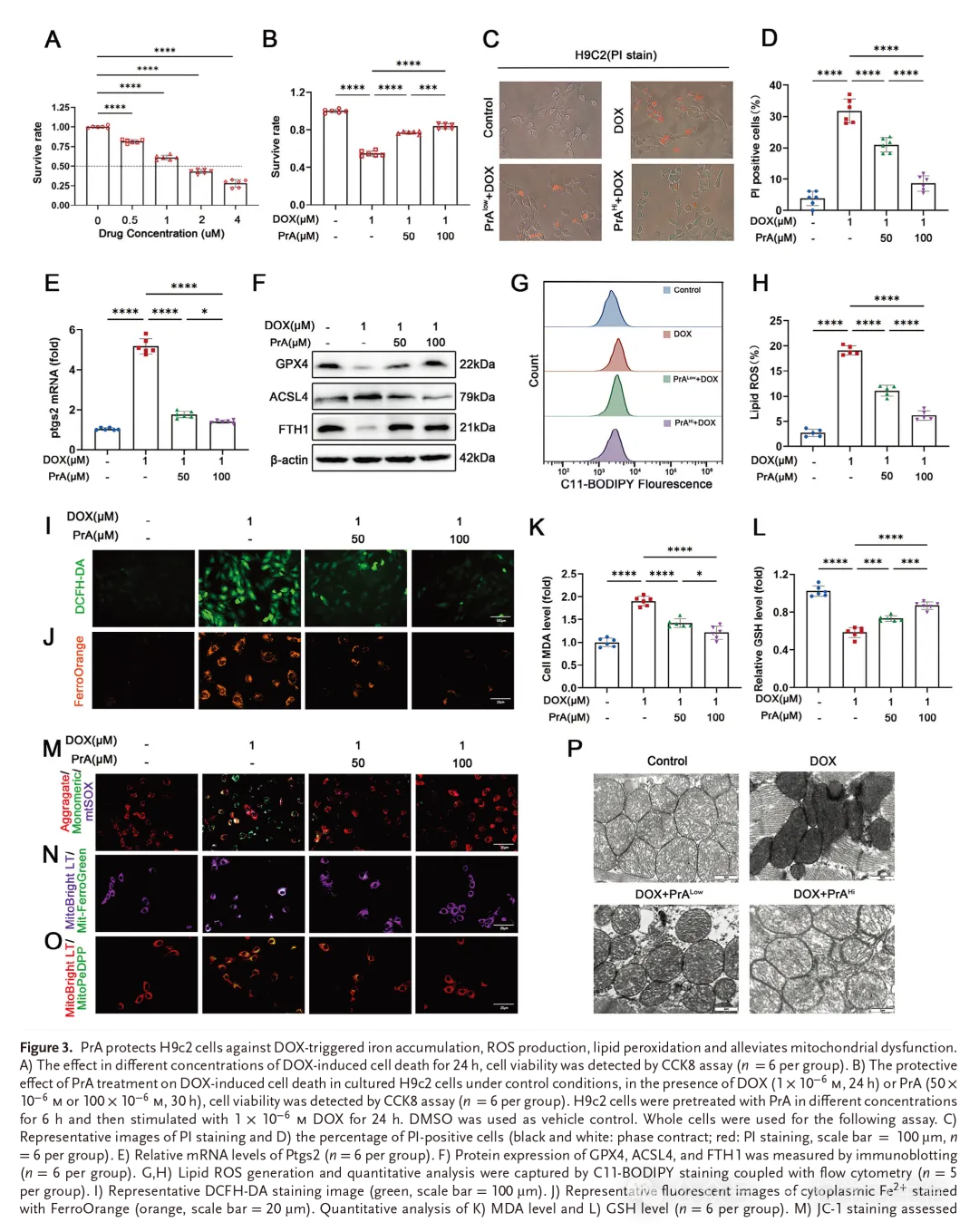
本研究探讨了PrA对大鼠H9c2体外细胞铁凋亡的细胞保护作用机制。将H9 C2细胞与不同浓度的DOX(0 × 10-6、0.5 × 10-6、1 × 10-6、2 × 10-6和4 × 10-6 μ M)一起培养24小时,导致细胞活力显著降低(图3A)。PrA处理对心肌细胞活力的影响几乎相同,在不存在DOX暴露的情况下,在50 × 10-6 m和100 × 10-6 m下无细胞毒性(图S3 A,支持性信息)。基于上述细胞存活率结果,我们选择了1 × 10-6 m DOX和50 × 10-6 m以及100 × 10?-6 mPrA的浓度用于后续实验。根据细胞计数试剂盒8(CCK8)和碘化丙啶(PI)荧光染色测定的结果,显示PrA在体外剂量依赖性地减少DOX诱导的心肌细胞损伤,证明了细胞保护作用(图3B-D)。与体内实验结果一致,PrA处理逆转了DOX条件心肌细胞中与铁沉积相关参数水平,包括Ptgs2基因(图3E)、GPX4、ACSL4和FTH1蛋白(图3F和图S3 B-D,支持信息),脂质和细胞内ROS水平(图3G-I和图S3 E,支持性信息)、细胞内亚铁离子(Fe 2+)含量(图3 J和图S3 F,支持性信息)和脂质过氧化产物MDA和GSH(图3 K,L)。这些结果表明PrA在体外有效地降低了DOX处理的心肌细胞对铁蛋白下垂的易感性。
线粒体依赖性铁积累和脂质过氧化是DOX诱导的铁细胞凋亡的关键因素。如前所述,我们发现,在DOX处理期间,培养的心肌细胞线粒体中的Fe2+和脂质过氧化物含量主要增加。然而,PrA处理消除了DOX诱导的这种现象(图3N,O和图S3H,I,J-M,支持性信息)。在上述所有条件下,我们进一步监测了反映线粒体功能的关键指标,包括线粒体膜电位(ΔΨm)和线粒体源性ROS(mitoROS)的产生。如图3M和图S3 G、J、K(支持性信息)所示,与对照组相比,DOX处理的心肌细胞表现出ΔΨm降低和线粒体超氧化物蓄积增加。相反,PrA干预逆转了DOX诱导的这些变化。类似地,在原代小鼠新生心肌细胞中也观察到PrA对DOX诱导的亚铁蛋白增多指数和线粒体损伤的保护作用(图S4,支持性信息)。电镜观察线粒体的结构和形态学特征。如图3P所示,揭示了DOX暴露导致线粒体收缩和线粒体膜密度增加,而PrA处理以剂量依赖性方式改善了小鼠心脏组织中DOX介导的异常特征。这些结果证明了PrA对DOX诱导的线粒体依赖性铁细胞凋亡的保护作用。
2.4.ACSL4和FTH1被鉴定为直接PrA结合靶点
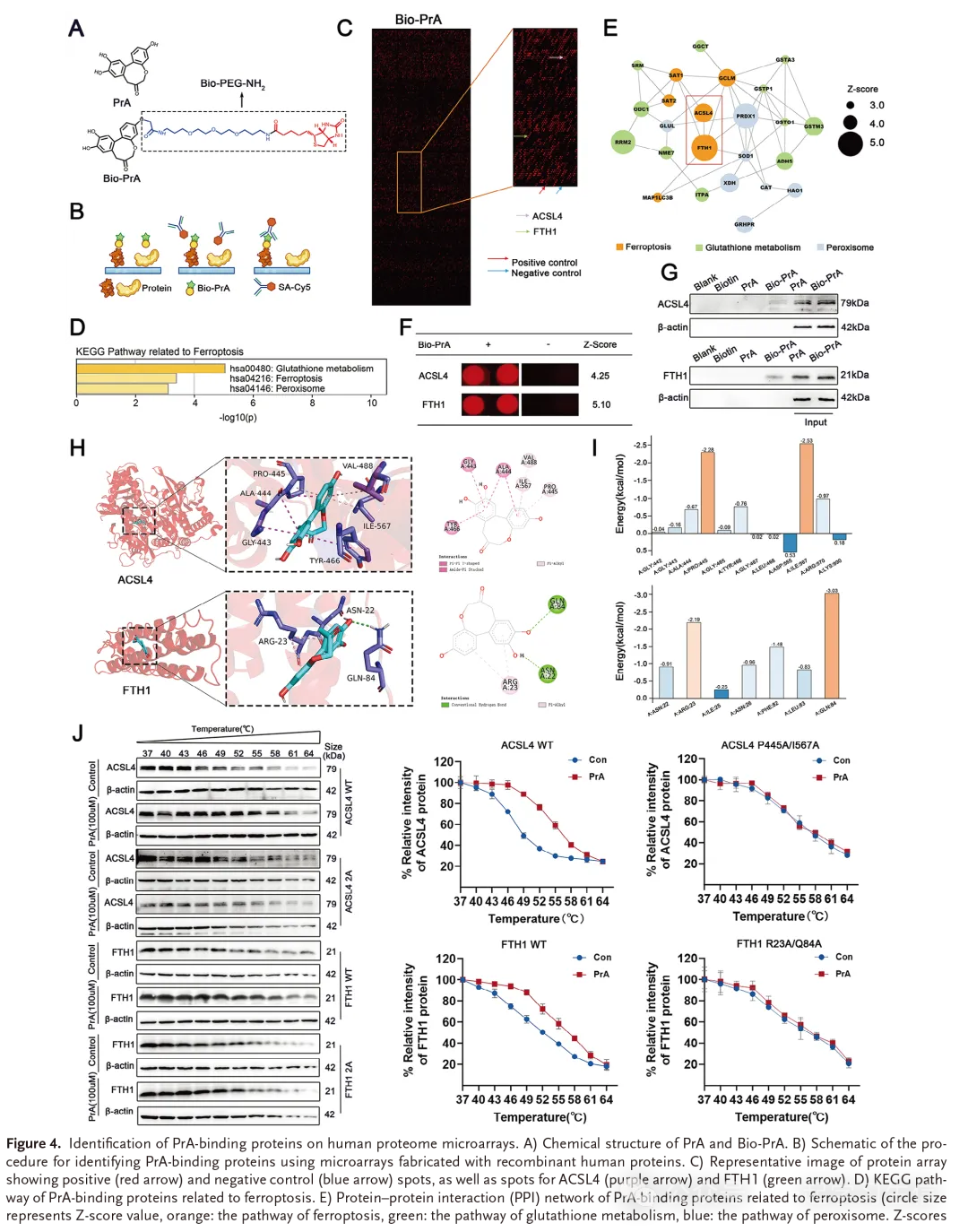
为了确定PrA抑制DOX介导的心肌细胞铁凋亡的潜在机制,将生物素标记的PrA(Bio-PrA;图4A)与Cy5缀合的链霉亲和素(Cy5-SA)串联使用,以通过检测PrA与在HuProt人蛋白微阵列上制备的重组蛋白的结合来鉴定PrA的潜在靶标(图4 B、C和图S5 A,支持信息)。KEGG分析显示,铁蛋白分解相关途径在PrA结合蛋白组中富集(图4D)。考虑到PrA在DOX介导的铁死亡中的重要作用,从Z分数中挑选出参与铁死亡途径的前两种蛋白(ACSL4和FTH1)进行进一步分析(图 4E,F)。
为了验证PrA是否与细胞裂解物中的ACSL4和FTH1结合,将Bio-PrA应用于链霉亲和素-琼脂糖珠并与H9c2细胞的裂解物共孵育。如下拉测定所示,Bio-PrA与来自H9c2细胞的裂解物中的ACSL4和FTH1蛋白结合(图4G)。随后,我们进行了分子对接和动力学模拟,以研究PrA如何与ACSL4和FTH1相互作用。对接结果表明,PrA与ACSL4通过α-羧基和α-烃基形成疏水相互作用,与FTH1通过常规氢键和α-烃基形成亲脂协同作用。所有这些相互作用有利于稳定结合复合物(图4H)。如动力学模拟所示,PrA和ACSL4之间具有最高两个平均能量值的关键残基是Ile567和Pro445,PrA和FTH1之间具有最高两个平均能量值的重要残基是Gln84和Arg23(图4 I)。为了验证分子对接和动力学模拟的结果,我们进行了细胞热位移试验(CETSA)。如图4J所示,PrA与ACSL4和FTH1物理相互作用,而在ACSL4的Ile567/Pro445和FTH1的Gln84/Arg23突变后,PrA与ACSL4或FTH1之间的相互作用明显受损。总之,PrA直接结合ACSL4和FTH1。
2.5.PrA通过抑制ACSL4磷酸化和激活抑制DOX诱导的铁凋亡
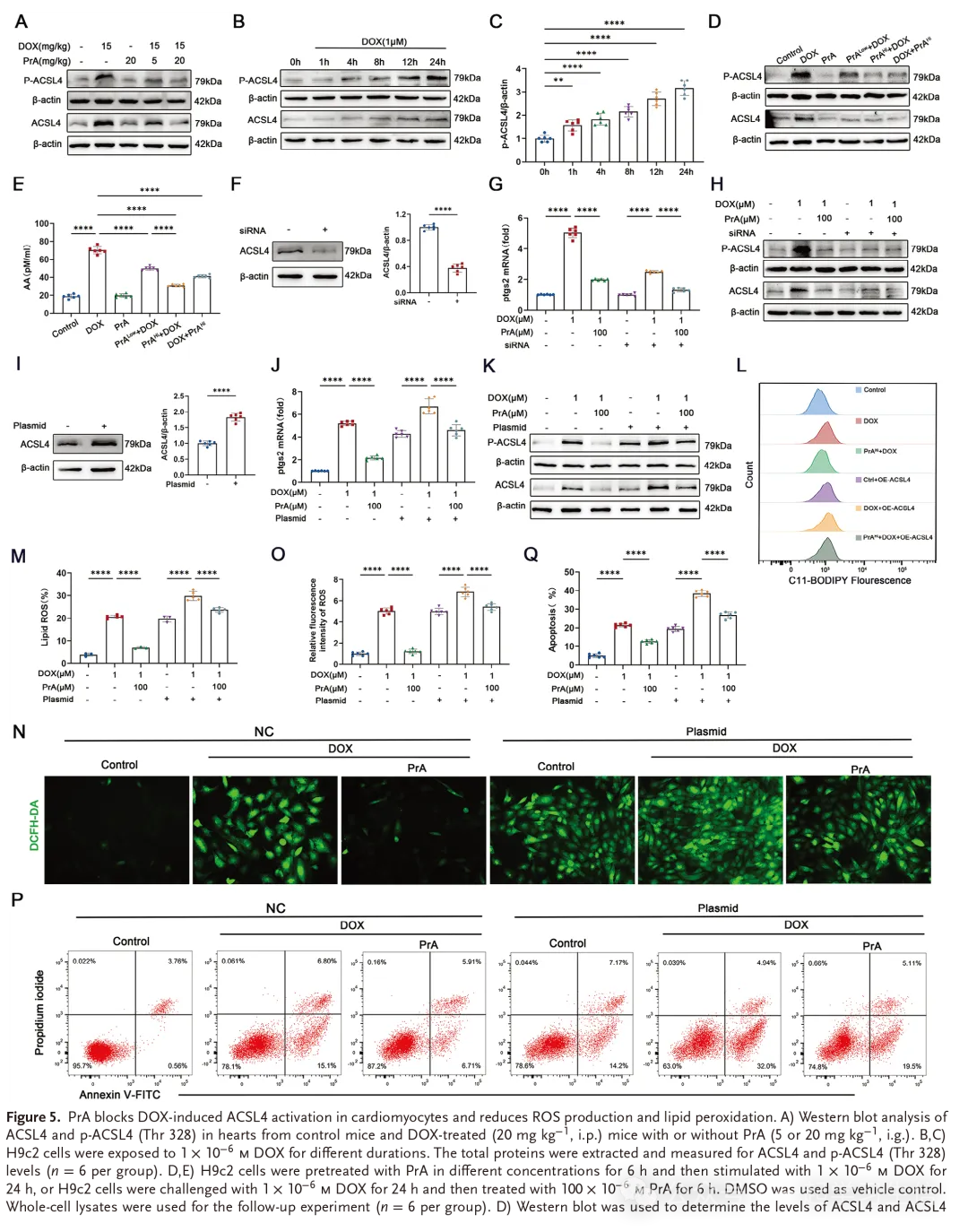
ACSL4是负责代谢多不饱和脂肪酸(PUFA)代谢的同工酶。最近的证据表明,Thr328磷酸化对于ACSL4酶促激活以增强铁凋亡敏感性是必不可少的。考虑到PrA直接与ACSL4结合,抑制DOX诱导的铁凋亡,我们研究了PrA是否通过干扰ACSL4的酶激活来保护DOX诱导的铁凋亡,Western blotting结果显示,DOX处理的小鼠心脏组织中ACSL4磷酸化和蛋白水平均上调,PrA以剂量依赖的方式逆转这些变化(图5A和图S6 A、B,支持信息)。随着时间的推移,在DOX处理的H9c2细胞中,ACSL4蛋白和Thr328磷酸化水平逐渐增加(图5B、C和图S6C,支持信息)。在DOX暴露24h后,高剂量PrA处理的H9c2细胞中ACSL4蛋白和Thr328磷酸化水平也受到抑制。值得注意的是,PrA预处理还降低了DOX条件化H9c2细胞中的ACSL4蛋白水平和Thr328磷酸化(图5D和图S6D,E,支持信息)。此外,单独的PrA处理不会影响小鼠心脏组织或来自对照组的H9c2细胞中的ACSL4蛋白或Thr328磷酸化水平。因此,在DOX存在下,PrA与ACSL4的结合干扰了ACSL4在Thr328的磷酸化及其蛋白表达。
ACSL4介导的PUFA代谢促进含花生四烯酸(AA)磷脂的合成和脂质过氧化,最终导致铁凋亡。因此,我们测量了在DOX存在下用或不用PrA处理的H9c2细胞中的AA水平。ELISA检测证实,单独的PrA预处理不改变AA的产生,但是,它逆转了H9c2细胞中细胞上清液中DOX介导的AA的产生。尽管DOX暴露后,高剂量的PrA保留了其拯救由D0OX引起的AA积累的能力(图5E)。基于这些结果,我们得出结论,PrA与ACSL4结合抑制了DOX诱导的ACSL4磷酸化和酶活性,进一步破坏了PUFA的下游代谢和脂质过氧化。
为了证实PrA是否通过阻止ACSL4磷酸化和活化来抑制DOX诱导的铁凋亡,在上述条件下,在用特异性靶向ACSL4的siRNA或过表达质粒转染的H9c2细胞中评价Ptgs2 mRNA水平。与PrA给药的数据一致,ACSL4沉默(图5 F)抑制了DOX条件化的H9c2细胞中的Ptgs2 mRNA水平,表明ACSL4敲低对DOX诱导的心肌细胞铁凋亡的抑制作用(图5G)。此外,ACSL4-siRNA和PrA对DOX诱导的铁凋亡的抑制作用与ACSL4 Thr328磷酸化减少相关(图5 H和图S6 F,G,支持信息)。相比之下,ACSL4过表达(图5I)促进DOX暴露的H9c2细胞中Ptgs2 mRNA表达(图5 J)和ACSL4 Thr328磷酸化水平(图5 K和图S6 H,支持信息)。在ACSL4饱和条件下,PrA对H9c2细胞中DOX介导的Ptgs2 mRNA(图5 J)、ACSL4蛋白和Thr328磷酸化(图5 K和图S6 H,I,支持信息)以及脂质和细胞内ROS水平(图5L-O)的扩增具有降低的补救能力。鉴于这些发现,进行流式细胞术,揭示ACSL4过表达损害了PrA对DOX诱导的心肌细胞死亡的保护作用(图5 P、Q)。综上所述,我们的数据表明,PrA通过阻止DOX诱导的ACSL4在Thr328磷酸化及其随后的激活来抑制心肌细胞铁凋亡。
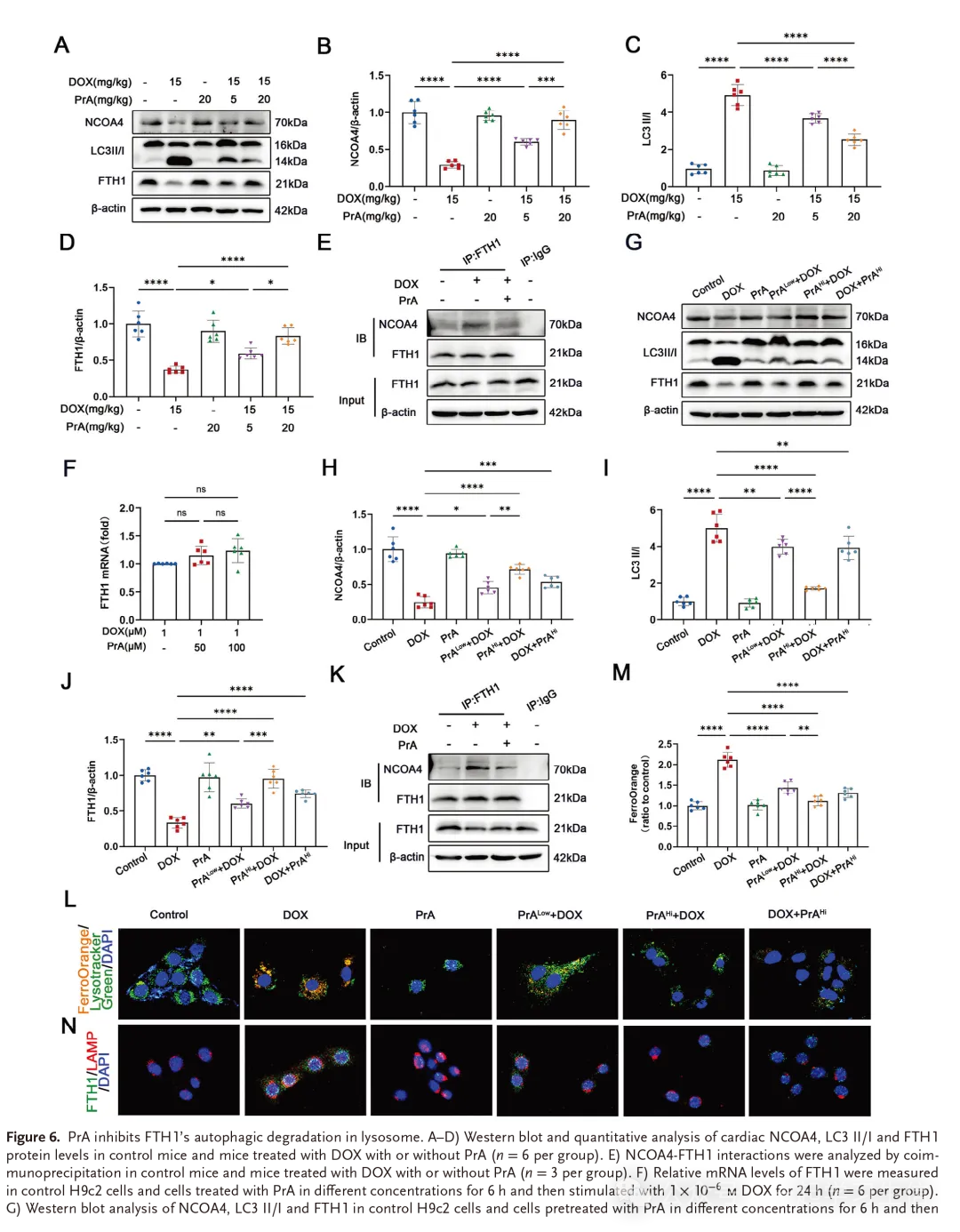
2.6.PrA通过减少溶酶体中FTH1的自噬降解保护DOX介导的铁凋亡
据报道,关键的铁蛋白亚基FTH1通过与货物受体NCOA4结合并将含铁的铁蛋白复合物转移到自噬溶酶体进行自噬降解来介导铁蛋白自噬,导致释放游离Fe2+并增加细胞对铁死亡的易感性。我们的数据显示,PrA可以直接结合FTH1,并恢复H9c2细胞中FOX诱导的FTH1蛋白水平(图3F)和细胞内Fe2+产生(图3 J)的改变。因此,我们推测PrA可能破坏DOX介导的NCOA4-FTH1相互作用,随后通过与FTH1结合来干扰铁蛋白吞噬。为了验证这种可能性,我们首先使用蛋白质印迹分析了FTH1,NCOA4和自噬标记LC3 II/I在DIC小鼠心脏治疗或不治疗PrA的蛋白质表达。与对照小鼠相比,DOX处理的小鼠在心脏组织中表现出较低的FTH1和NC0A4表达以及较高的LC3-II/I百分比,而PrA干预逆转了这些变化(图6A-D)。免疫共沉淀测定进一步显示,在DOX处理的小鼠中,PrA给药减弱了FTH1和NCOA4之间的相互作用(图6 E)。与体内结果一致,PrA恢复了H9c2细胞中由DOX引起的FTH1蛋白水平而不是mRNA表达(图6 F),表明PrA通过抑制蛋白降解而不是通过上调基因转录来调节DOX存在下的FTH1蛋白水平。类似地,在DOX暴露之前和之后,PrA挽救了H9c2细胞中NC0A4的蛋白表达和LC3-II/I百分比,以及NC0A4-FTH1相互作用,而单独的PrA处理对这些指数没有影响(图6 G-K)。因此,我们的数据支持PrA和FTH1的组合抑制DOX触发的NCOA4-FTH1内部相互作用和FTH1蛋白自噬降解的结论。
为了阐明PrA预防DOX诱导的铁自噬的机制,我们使用特异性荧光探针追踪Fe2+和溶酶体来评估细胞内Fe2+含量和亚细胞位置。激光共聚焦显微镜显示,DOX处理的H9c2细胞在溶酶体中发射强的Fe2+荧光,其通过在DOX暴露之前施用的增加剂量的PrA而减弱,表明PrA抑制了DOX诱导的细胞内Fe2+从溶酶体释放到细胞质中(图6L,M)。根据免疫荧光测定,DOX处理加速FTH1向溶酶体的移位。相反,在PrA作用下,FTH1和溶酶体的共定位被消除(图6 N)。尽管DOX处理后,高剂量PrA仍然抑制FTH1和溶酶体的共定位,以及Fe2+释放到细胞质中(图6L-N)。总的来说,PrA和FTH1的结合破坏了NCOA4-FTH1的相互作用,抑制了铁蛋白吞噬和溶酶体中Fe2+的释放,从而改善了DOX诱导的心肌细胞铁凋亡。
2.7.PrA介导的铁凋亡抑制减轻缺血再灌注诱导的心脏损伤和功能障碍
最近的研究表明,铁凋亡和心肌I/R之间有很强的相关性,这表明铁凋亡干预可能会在改善再灌注后的心脏功能方面提供治疗益处。因此,我们测试了PrA治疗是否对心肌I/R损伤具有治疗作用。建立心肌I/R鼠模型,并用或不用PrA(20mg/kg)处理指定天数(图7A)。与I/R小鼠相比,PrA治疗部分恢复了I/R诱导的血清心肌损伤标志物活性升高,包括CK-MB和LDH(图7 B,C)。如图7 D和图S7 A(支持性信息)所示,PrA预处理组的梗死面积显著低于I/R组。随后,我们使用超声心动图评估心脏功能。I/R诱导的心功能不全主要表现为LVEF、FS、LVIDd和LVID的变化,这些变化在PrA治疗后恢复(图7 E-I)。此外,组织学染色显示PrA显著改善I/R诱导的心脏纤维化(图7 J)。
为探讨PrA对心肌缺血再灌注损伤中铁凋亡的保护作用机制,我们采用PCR方法检测了缺血再灌注小鼠心肌中Ptsg2 mRNA的表达水平。如预期的,在PrA处理的I/R小鼠中Ptgs2 mRNA水平显著下调(图7 K)。此外,I/R组的心脏和血清MDA水平高于PrA治疗组(图7 L,M)。PrA处理恢复了I/R诱导的小鼠心脏组织中GPX4、ACSL4和FTH1蛋白(图7 N和图S7 B-D,支持性信息)、铁(图7 O和图S7 E,(支持性信息))和ROS含量(图7 P和图S7 F,支持性信息)的变化。我们的研究结果表明,PrA对心肌I/R损伤具有抗铁凋亡和心脏保护作用。

3. Discussion
目前,对于化疗药物引起的心血管毒性反应的防治尚无共识。现有的缓解心脏毒性的策略主要涉及调整药物递送模式和剂型设计,或给予心脏保护药物,而其临床应用和开发通常由于副作用增加、体内活性差和成本高而受到限制。因此,有必要探索切实可行的干预措施,以减轻化疗引起的心脏毒性。我们的研究首次证明 PrA在小鼠DIC模型中以剂量依赖性方式防止DOX诱导的心脏损伤和功能障碍。此外,在改善DOX诱导的心肌损伤方面,20mg/kg PrA介导的保护作用部分上级DXZ,显示出其在治疗DIC方面潜在的实际和临床应用价值。
心肌细胞死亡是DOX所致心肌损伤和心功能不全的病理特征。新出现的细胞死亡形式-铁细胞凋亡,是癌症、缺血性器官损伤和其他与脂质过氧化密切相关的退行性疾病的主要致病机制之一。大量的证据表明,抑制心肌细胞铁蛋白增多是一种有前途的预防性治疗DIC的策略。然而,PrA是否调节心肌细胞中DOX介导的铁细胞增多仍不清楚。近年来的研究表明,铁蛋白缺乏主要由外源性(转运蛋白依赖性)和内源性(酶调节性)途径诱导。此外,脂质代谢失衡、铁过载和氧化应激已被确定为铁下垂症的诱发因素。在此基础上,我们的研究表明,PrA可以通过逆转DOX诱导的GPX4、FTH1、ACSL4、Fe2+、脂质过氧化产物和ROS水平的变化,通过上述内源性和外源性途径减轻DOX诱导的心肌组织和心肌细胞铁蛋白增多。
越来越多的证据表明,铁细胞增多症和线粒体信号传导之间存在密切联系。细胞铁蛋白缺乏导致线粒体结构和功能的不规则性,进一步加剧了细胞对铁蛋白缺乏的易感性,并形成了正反馈环。研究表明,线粒体疾病通过ROS生成、谷氨酰胺分解代谢和铁代谢驱动铁细胞增多症。功能完整的线粒体主要通过促进脂肪酸β-氧化、抑制GPX 4催化活性和脂质过氧化作用而获得抗铁下垂的能力。在另一项近期研究中,DOX调控的心脏cGAS-STING通路引发了线粒体功能障碍,这是心肌损伤和心功能不全不可或缺的机制。为了支持这些发现,我们通过监测亚铁蛋白下垂、线粒体形态和功能的相关指标,观察到PrA以线粒体依赖性方式改善了DOX诱导的心肌细胞亚铁蛋白下垂。
ACSL4是驱动铁死亡的内源性途径中的关键同工酶,可协同促进PUFA掺入磷脂中,形成含PUFA的磷脂,磷脂易受脂氧合酶引发的氧化反应的影响,破坏脂质双层并损害膜功能。ACSL4的促铁下垂作用和潜在致病作用最近已被映射到各种多系统疾病,包括辐射诱导的肠损伤、脑缺血/再灌注和心脏微血管损伤。尽管有上述丰富的证据,但关于ACSL4在DIC中的作用机制的研究很少。有研究报道,在DOX诱导的铁凋亡过程中,ACSL4蛋白在心肌细胞中上调。然而,DOX在DIC中调节ACSL4的机制仍然未知。最近的一项研究表明,ACSL4上的Thr328被PKCβ II磷酸化形成二聚体复合物,进一步激活其酶促作用,驱动脂质过氧化和铁凋亡。通过筛选与PrA结合的潜在下游靶蛋白,我们发现PrA主要通过Ile 567/Pro 445残基与ACSL4物理结合,表明PrA通过直接靶向铁凋亡途径保护DOX诱导的心脏毒性。结合PrA在DOX激发条件下抑制ACSL4蛋白表达的结果,我们推测PrA通过干扰ACSL4的酶活性来抑制DOX诱导的心肌细胞铁蛋白增多。基于一系列的体内和体外实验,我们首次证明DOX暴露上调了心肌细胞和心脏组织中ACSL4的磷酸化和蛋白表达,激活了其酶促反应,随后脂质过氧化产物聚集,增加了对铁细胞增多症的易感性。相反,PrA在DOX处理之前和之后都有效地恢复DOX介导的铁细胞凋亡,这表明PrA通过结合ACSL4并抑制其酶活性来减轻DOX诱导的铁细胞凋亡。
铁转运和超负荷是外源性铁凋亡的基本机制。在正常条件下,铁储存在铁蛋白(由铁蛋白轻链和FTH 1组成)中,并且铁转运蛋白通过促进铁的细胞摄取来维持铁平衡。在细胞内铁缺乏的情况下,铁蛋白与自噬受体结合形成复合物,进一步连接到LC 3-II并运输到自噬体进行降解,允许铁蛋白结合的铁释放到细胞质中,这一过程称为铁自噬。研究表明,细胞质自噬受体NCOA 4选择性识别铁蛋白中的FTH 1亚基,并且FTH 1 R23是铁蛋白结合NCOA 4所必需的有影响的结构。干扰FTH 1和NCOA 4之间的相互作用可以防止铁自噬,并进一步降低线粒体铁含量,从而在肺纤维化,青光眼和神经退行性疾病中发挥关键的治疗作用。在我们的研究中,使用分子对接和热转移实验将FTH1鉴定为PrA的下游结合蛋白。在此基础上,我们探讨了PrA和FTH1在DOX诱导的铁细胞凋亡中的相互作用机制。在我们的体内和体外实验中,PrA挽救了DOX介导的心脏组织和心肌细胞中FTH1、NCOA4和LC3-II/I蛋白水平的改变以及FTH1-NCOA4相互作用。结合PCR和Western blot分析表明,PrA在转录后水平调节DOX条件下的FTH1表达,表明PrA可能在DOX存在下调节FTH1介导的铁自噬。使用特异性荧光探针追踪细胞内Fe2+和溶酶体进一步验证了这一假设。免疫荧光和共聚焦显微镜显示,PrA阻止FTH1-溶酶体共定位和释放到细胞质中的Fe2+在DOX的存在下。因此,我们得出结论,PrA结合FTH1,竞争性抑制FTH1-NCOA4相互作用,阻断FTH1介导的铁自噬,并保护心肌细胞免于铁凋亡。
近年来的研究表明,铁凋亡是心肌I/R损伤的重要病因,抑制铁凋亡可有效减少I/R引起的心肌细胞死亡,改善左室收缩功能。为了探讨PrA作为铁凋亡抑制剂在临床应用和转化开发中的可能性,我们分析了PrA对心肌I/R损伤的影响。值得注意的是,我们的结果证实了PrA在小鼠心肌I/R模型中的心脏保护和抗铁毒性作用。在后续的实验中,我们将进一步验证PrA调节AC16心肌细胞铁凋亡的具体机制。此外,PrA在其他铁中毒介导的疾病中的治疗作用和抗铁中毒活性也值得进一步研究。此外,我们打算进行一系列的临床试验,以验证PrA治疗药物介导的心脏毒性的安全性和有效性。
4. Conclusion
我们的研究首次揭示了PrA通过抑制ACSL4/FTH1依赖性铁凋亡途径来减轻DOX诱导的心肌损伤。此外,在心肌I/R小鼠模型中,阐明了PrA的心脏保护和抗铁毒性作用。总的来说,我们的数据表明,PrA可以作为一种新的双重铁蛋白分解靶向抑制剂,具有很好的进展和转化价值,为DIC和其他铁蛋白分解介导的疾病的潜在治疗途径。
5. Experimental Section
药物:PrA(JOT-10793,纯度≥98%)购自成都普菲德生物技术有限公司(中国成都)。阿霉素(DOX,D107159,纯度≥98%)购自Aladdin(中国上海)。盐酸右雷佐生(DXZ,ab141109,纯度≥98%)购自Abcam(中国上海)。本研究中使用的抗体列于表S2(支持性信息)中。
小鼠:野生型C57BL/6小鼠(6-8周龄,20 ± 2 g)购自北京市实验动物研究中心(中国北京)。将小鼠圈养在温度受控的环境(22-24°C)中,光照-黑暗周期为12小时,并喂食标准啮齿类动物实验室饲料。所有实验方法和方案均由哈尔滨医科大学附属第二医院动物护理和使用委员会(编号Sydwgzr 2020-014)根据ARRIVE指导原则批准。鉴于C57BL/6雄性小鼠常用于建立DIC模型,本研究中主要使用雄性动物。然而,考虑到DOX诱导心脏毒性的显著性别差异,进一步纳入雌性C57BL/6小鼠进行验证。
多柔比星诱导的急性心脏损伤模型:对于急性实验,6-8周龄小鼠接受单次腹膜内(i. p.)注射DOX(15mg/kg体重溶于无菌盐水)或盐水。如有指示,小鼠接受PrA预处理(5mg/kg或20mg/kg)经口灌胃或DXZ(25mg/kg),然后在DOX处理后2和48h,用相同体积的媒介物连续接受相同的处理再两次(Veh; 10%二甲亚砜DMSO与磷酸盐缓冲盐水PBS)作为对照腹膜内施用。将PrA和DXZ溶于10%DMSO中,并在无菌盐水中稀释。
I/R心肌损伤模型:将成年雄性小鼠(6-8周龄)用三溴乙醇(M2910,AiBei Biotechnology,Nanjing,China,20ml/kg,i. p.)并以仰卧位放置在实验板上。镇静后,使用小动物呼吸机对小鼠进行插管。在进行左胸廓切开术切口后,通过使用带有活结的7-0丝线可逆性结扎左前降支(LAD)冠状动脉来诱导缺血。通过目视观察左心室壁变白确认结扎正确。局部缺血30分钟后,活结被释放,导致结扎远端变色心肌恢复。PrA(20mg/kg)在手术前24和2小时通过口服灌胃给药。假手术对照小鼠(假MI/R)进行相同的外科手术,但没有结扎LAD冠状动脉。
细胞培养:如前所述分离新生原代心肌细胞。H9c2和HEK-293细胞获自中国科学院细胞库(中国上海)。将细胞培养在补充有10%胎牛血清(FBS,Pricella)和1×青霉素-链霉素(Beyotime Biotechnology,Shanghai,China)的Dulbecco改良Eagle培养基(DMEM,Gibco)中,并在37°C下在由5% CO2和95%空气组成的潮湿气氛中孵育。将DOX以指定浓度应用于H9c2细胞和原代心肌细胞24小时。在DOX治疗前或治疗后6小时,以指定浓度进行PrA干预。
蛋白质组微阵列分析:由Wayen Biotechnology(中国上海)使用含有23145种纯化人蛋白的HuProt 20K蛋白质组微阵列鉴定PrA相互作用蛋白。简言之,将微阵列在室温(RT,20°C)下浸入封闭缓冲液(PBS-T中的5%牛血清白蛋白)中1小时。然后将微阵列与10×10-6m生物素化PrA或游离生物素孵育1h。洗涤后,将蛋白质组微阵列在室温下在黑暗中置于0.1%Cy5-链霉亲和素溶液中20分钟。再次洗涤并离心(1000×g,2min)至干燥后,使用GenePix 4000B微阵列扫描仪(Axon Instruments,美国)和GenePix TM Pro v6.0软件(Axon Instruments,美国)进行观察和数据分析。在PrA-Bio处理的微阵列中Z-Score(信号强度指标)≥2.8和在生物素处理的微阵列中Z-Score<2.8的蛋白质点被鉴定为候选阳性蛋白。将PrA-Bio处理与生物素处理的Z评分比定义为I平均值比,将I平均值比≥1.4的候选阳性蛋白确定为最终PrA靶蛋白。
统计分析:使用GraphPad Prism软件(9.0版,GraphPad Software,Boston,MA,USA)分析数据并绘图。各统计分析的生物学重复(n)见图例。使用Kaplan-Meier分析和对数秩(Mantel-Cox)检验构建生存曲线。所有汇总数据均以均值±标准差表示,并使用两组之间的双尾未配对学生t检验或多组之间具有Tukey多重比较的单因素方差分析进行评估。在p<0.05时,差异被认为具有统计学意义。