大家好,我是带我去滑雪!
因果学习与生物医学的交叉融合 是近年来人工智能在生命科学领域的一个关键突破方向,展现出巨大的应用潜力和科研价值。传统的生物医学研究往往依赖相关性分析来揭示疾病机制、筛选药物靶点或评估治疗效果,但仅有相关性并不能确保干预的有效性与安全性。因此,因果学习的引入为生物医学提供了一种能够推断干预结果、揭示潜在机制的新范式,尤其在精准医疗、药物开发和复杂疾病研究中展现出独特优势。在药物研发方面,因果模型可以帮助识别药物与表型之间的因果关系,加速药物再定位过程并降低临床试验成本。在医疗决策支持中,因果推断能基于电子病历推测特定治疗对个体的真实效果,从而实现个性化治疗推荐。在疾病机制研究方面,因果图模型可揭示基因调控网络、分子通路中变量之间的方向性关系,为疾病靶点发现提供理论依据。尽管如此,该领域仍面临挑战,包括生物医学数据中混杂因素众多、数据质量参差不齐、因果结构难以验证等问题。此外,**++如何将深度学习与因果推理有效结合,实现多模态高维数据的因果建模,也是当前研究热点之一。++**未来,随着因果推理理论的进一步发展及其与临床实践的深入结合,其在疾病预测、早筛机制、治疗优化等方面的应用将日趋广泛,成为推动医学智能化的重要引擎。
今天分享一篇关于因果学习与生物医学交叉领域的科研论文,题为《Identifying cancer prognosis genes through causal learning》,发表期刊为Briefings in Bioinformatics。
目录
(1)通过CPCG鉴定的致病基因在生存时间上表现出很强的区分力
一、论文概述
精准识别癌症预后关键基因对于指导肿瘤治疗至关重要。由于混杂偏差、选择性偏差等,使得传统基于广义相关性的特征选择方法识别出的生物标志物的预测性能常常不稳定。加之临床数据常伴随样本量少、删失数据多等情况,进一步加剧了有效生物标志物识别的挑战,相较而言,因果特征的稳定性在这些方面具有显著优势。针对上述情况,本研究提出新的计算框架CPCG ,++通过集成参数和半参数比例风险模型评估基因表达对患者预后的影响,确定候选基因集合;随后通过因果关系推断构建候选基因与患者总生存时间关系的因果骨架,最后与总生存时间直接相关的一阶邻居基因作为癌症预后的核心基因。++实验结果表明,CPCG能够显著消除基因与患者预后之间的虚假关联,直接从高维转录组数据中识别出一组简洁可靠、具有可解释性、泛化性和稳健性的癌症预后关键基因。CPCG也为其他医学任务的稳健生物标志物识别提供参考。
二、主要创新点
在这项研究中,提出了CPCG(癌症预后的因果基因),一个两阶段的框架,用于确定预后的因果基因。在第一阶段,CPCG旨在获得与预后显著相关的简化基因数据集,以便于随后的因果骨架构建。采用集成策略,使用参数和半参数风险模型,以模拟基因表达和患者预后之间的关联。在第二阶段,利用这些基因与总生存时间作为目标变量。通过对每个变量对进行迭代条件独立性检验,大致构建了一个因果框架,选择与总生存时间直接相关的基因作为预后因果基因。为了评估预测性能,使用各种指标并验证不同数据集的通用性。此外还探讨了这些基因的生物学功能。
三、材料与方法
CPCG用于肿瘤预后基因识别的框架在第一阶段,目标是通过使用参数和半参数策略筛选候选基因,重点是风险作为目标变量。在每种策略中,和
代表不同总体和模型计算的风险,第一阶段的
由指数比例风险模型进行转换。对数秩检验(
)的P值用于筛选不相关基因。在第二阶段,CPCG通过迭代的条件独立测试和图修剪从候选基因中识别与癌症预后相关的致病基因。 CPCG的流程框架如下图所示
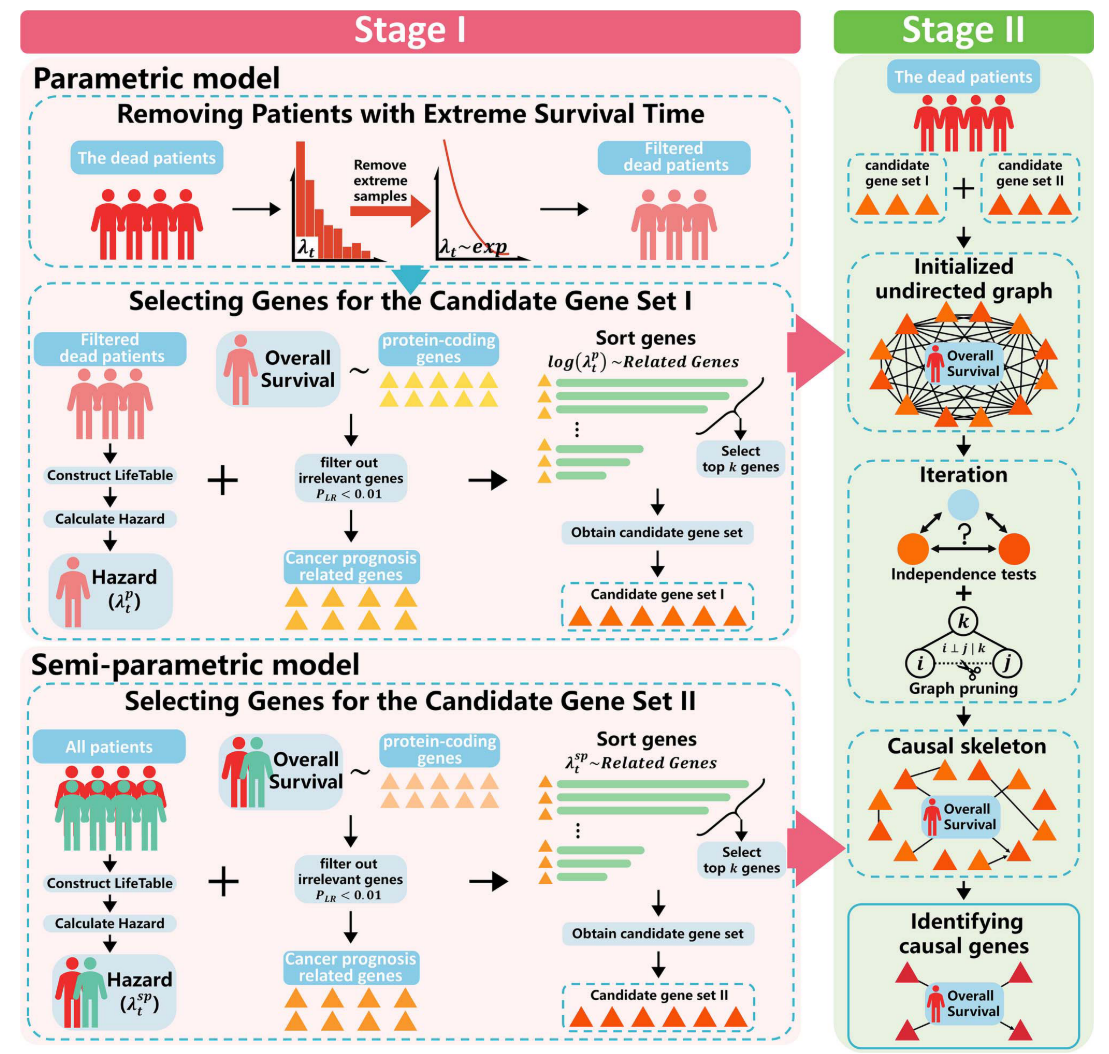
在CPCG方法的第一阶段中,参数模型特别擅长精确量化基因表达水平对癌症预后的影响。该模型假设患者的风险符合指数分布,这允许在特定框架内详细和准确地评估基因影响。另一方面,半参数模型在跨更广泛的患者数据范围外推候选基因方面表现出色,而不依赖于这种具体的分布假设。这种灵活性使得能够识别可能不符合参数模型的严格假设的基因,从而拓宽了潜在预后标记物的范围。通过结合来自两个模型的候选基因集,产生一个更全面的基因列表。该列表包括可由指数比例风险模型解释的基因,以及不受数据分布假设约束的基因。因此,通过CPCG识别的基因表现出更高的统计鲁棒性,并可适应不同的数据集。此外,这种协作方法有助于识别在使用单一方法时可能被忽视的基因。
在CPCG的第二阶段中,将基因集I和基因集II结合起来,形成一个候选基因库,用于进一步鉴定影响癌症预后的致病基因。利用已故患者的数据,采用条件独立性测试和图修剪技术,以构建一个因果骨架,旨在确定从候选池的因果基因。最初,所有的变量对,包括候选基因和生存时间,连接形成一个无向图。随后,我们进行联合边际独立性检验和条件独立性检验(条件独立性检验的描述见补充方法),以部分确定每对之间的直接关系。然后删除变量之间仅显示条件依赖的边缘。我们根据变量间的偏相关系数来确定变量间的条件独立性
四、数据收集与预处理
从癌症基因组图谱计划(TCGA)收集数据,该计划涉及全身18种不同癌症类型的7371名患者(图2A),包括总共2654例(36%)死亡病例和4717例(64%)存活病例。具体来说,主要关注TCGA数据集中死亡人数超过100人的癌症类型。设定该阈值是为了确保进行的统计分析稳健且有意义。除了这些数据集,还包括ESCA,UCEC,LAML,MESO和SARC等癌症类型。选择这些国家是因为它们的中等死亡率人口,死亡人数在50至100人之间。纳入这些数据集使其能够评估本文的CPCG方法在各种死亡率量表中的普遍性和有效性。为了确保所识别的因果基因的可靠性和临床相关性,本文只关注蛋白质编码基因,并过滤掉平均原始计数低于10的基因。用于每种癌症的基因数量在图2A中详细描述。在数据预处理中,本文使用每百万转录本(TPM)标准化来标准化转录组数据。
为了评估本文的方法的通用性和鲁棒性,本文收集了来自基因表达综合数据库(GEO)中的12种癌症类型的另外24个独立数据集和来自中国神经胶质瘤基因组图谱计划(CGGA)中的1种癌症类型的2个数据集(图2B)。这些数据集包括4625例患者,其中2103例(45%)死亡病例和2522例(55%)生存病例。在这里,从这两个数据库中,本文发现了6个死亡人数超过100人的数据集,另外6个数据集包含中等数量的死亡病例,此外,还有12个死亡人数<50人的数据集。我们希望这些常见的癌症数据集可以验证CPCG识别的基因与大,中,小死亡病例人群癌症预后之间的关系。来自GEO的基因表达水平存储在微阵列数据中,并且我们对数据应用log2变换。通过高通量转录组测序获得的CGGA数据使用TPM标准化进行标准化。
五、评价指标
使用四个主要指标来评估CPCG的预后预测性能:
- 对数秩检验(PLR)的P值:对数秩检验是用于比较两组生存时间分布的标准统计方法。所得P值可以指示来自不同基因表达水平的组的患者之间观察到的存活差异的统计学显著性,并评估基因对患者预后的影响,P值越小,表明差异越显著。
- 考克斯回归的一致性指数(C指数):C指数测量通过考克斯模型估计的患者预测预后与实际事件发生之间的一致性。较高的C指数表明考克斯模型在患者风险方面具有更好的预测能力。
- 在对数秩检验(PSPL)中具有显著P值的基因比例:鉴于CPCG和COX-LASSO之间输出基因数量的显著差异,基于对数秩检验中的所有P值直接比较其性能具有挑战性。因此,PSPL被设计为全面地表示来自两种方法的具有显著P值的变量的比例,其中较高的PSPL指示存在来自相同集合的具有显著P值的更多基因对患者预后有很大影响。这有助于直接比较已确定变量的重要性。
- 考克斯回归(PSPC)中显著基因的比例:一个稳健的回归模型应该包含与目标变量真正相关的变量,并在回归中表现出显著的P值,同时避免多重共线性问题。在回归变量中,较高比例的显著变量表明所选预测因子的可靠性。因此,PSPC代表考克斯回归模型中显著影响预测的肿瘤相关基因中的存活时间的自变量(鉴定的肿瘤相关基因)的比例。更高的PSPC意味着考克斯模型包含更多真正影响患者生存时间的基因,并且这些基因在预后预测中起重要作用。
六、实验设置
在第一阶段,我们在对数秩检验中建立了P值阈值0.01,并过滤掉P值超过0.01的基因。对于参数和半参数模型,我们选择前100个基因作为候选的肿瘤相关基因。进入第二阶段,我们利用Fisher Z检验来评估条件独立性检验的显著性,α设置为0.05。
七、实验结果
(1)通过CPCG鉴定的致病基因在生存时间上表现出很强的区分力
(2)CPCG在其他独立测试数据上表现良好
(3)已鉴定致病基因的生物学相关性
(4)CPCG对数据输入顺序的敏感性较低
(5)CPCG不需要考虑Markov等价类和V-结构
八、讨论与结论
提出了一个两阶段的框架CPCG从因果关系的角度来识别与癌症预后相关的基因,因为因果关系比相关性更稳定。实验表明,通过CPCG获得的基因在癌症预后预测中表现出稳定性、可靠性、良好的可重用性和增强的可解释性,有助于更有效地干预疾病。在进行因果推理之前,对初步筛选的因果基因,集成参数和半参数成分进行风险处理,构建每个基因与癌症预后的一一对应关系,有助于精确缩小潜在因果基因的集合规模,避免高维特征对相关性度量的影响,无疑有利于后续因果特征选择的效率和有效性。这是因为在高维但相对低样本的情况下因为它在计算上可能是复杂的,并且在某些情况下,甚至是棘手的。因此,对候选基因进行精确的初步筛选至关重要。
CPCG第二阶段中使用的条件独立性检验可以有效地解决多重共线性问题。我们还证明了在结构学习模块中,CPCG对数据的输入顺序不敏感,这也归因于从阶段I获得的因果基因的精确选择。此外,我们表明,我们的方法不需要考虑马尔可夫等价类,这是一个具有挑战性的问题,在因果结构学习。这些都使得使用CPCG涉及较少的考虑。我们的方法无疑消除了基因和生存之间的虚假关系,其中一些是多重介导变量传递后的效应,一些是由于常见的原因。因此,它有助于肿瘤系统生物学家和临床医生解决长期存在的问题。传统的基于特征选择的生物标志物识别方法在实际临床应用中也面临一定的差距,其中之一就是如何选择最终的小集合以获得最佳的预测性能。通常,最佳集合是通过枚举获得的。我们的方法可以获得一个小的因果基因集,而不需要进一步耗时的枚举。
通过文献综述,它还表明了已确定的致病基因与癌症进展和恶性程度之间的生物学关联。此外,我们调查并发现68个基因中有13个与分泌蛋白有关,其中11个基因编码的蛋白在血液中容易检测到(CYP2E1、H2AW、CD5、OAF、CBR1、CRTAC1、RARS1、KIRP3、CYP2C18、SNAP25、CD33)68 - 70;这些进一步强调了这些致病基因作为临床预后生物标志物的潜力,即它们编码的蛋白质可以在癌症患者的体液中检测到。我们的方法也有进一步改进的余地。目前,CPCG仅关注转录组学数据;如果其他多组学数据以及成像数据成为更大的样本量,我们将继续探索与其他特征相关的因果关系,
参考资料
Siwei Wu, Chaoyi Yin, Yuezhu Wang, Huiyan Sun, Identifying cancer prognosis genes through causal learning, Briefings in Bioinformatics, Volume 26, Issue 1, January 2025, bbae721.
更多优质内容持续发布中,请移步主页查看。
点赞+关注,下次不迷路!