
综述论文
● 期刊:aBIOTECH (IF:5.0)
● 文章链接::https://www.sciencedirect.com/science/article/pii/S2662173825002115
● 第一作者:罗豪、王瑶
● 通讯作者:刘永鑫、杨军波
● 合作作者:侯辉宇
● 发表日期:2025-10
● 主要单位:
中国农业科学院深圳农业基因组研究所
摘要
传染病的持续出现迫切需要前沿的诊断方法。传统的病原体诊断手段受限于检测范围窄、处理周期长以及灵敏度不足等问题。高通量测序技术,尤其是基于多重聚合酶链式反应(PCR)的靶向测序,已成为病原体检测领域的变革性工具,具备更高的灵敏度、特异性及成本效益。然而,诸如引物二聚化及扩增偏倚等引物设计问题仍限制了此类方法的检测效率。本综述系统探讨了测序技术的最新进展,重点阐述了培养组学、宏基因组学及宏转录组学在病原体发现中的重要作用。我们进一步介绍了新型的容错引物设计策略,该策略通过在覆盖度与特异性之间取得平衡,有效优化多重PCR体系。此外,人工智能技术的引入显著提升了测序的精确性与可扩展性,使实时诊断成为可能。总体而言,这些进展为构建稳健而可持续的病原体检测体系提供了新的方向,对促进全球健康、粮食安全与生态系统韧性具有重要意义。
引言
畜牧业因动物传染病而经常遭受严重的经济损失,近年典型的病例包括高致病性禽流感、非洲猪瘟以及口蹄疫等。同时,人畜共患病原体对食品安全与公共健康构成重大威胁。世界卫生组织的统计显示,全球约60%的新发传染病及75%的新出现的人类病原体均源自动物。典型的人畜共患病原体包括沙门菌属、弯曲菌属、布鲁氏菌属以及β-冠状病毒属的SARS-CoV-2,这些病原体均曾引发严重的公共卫生事件。由SARS-CoV-2导致的新冠疫情凸显了人畜共患病原体从动物宿主(如蝙蝠或其他中间宿主)中溢出并迅速演化为全球性健康危机的巨大潜能。该疫情不仅造成数百万人死亡,还以前所未有的规模破坏了全球经济、粮食体系与公共卫生基础设施。
与动物病原体相似,植物病原体同样对农业生产力与生物多样性造成了巨大损失。例如,晚疫病菌导致马铃薯晚疫病,快疫木质杆菌引起橄榄树快速衰退综合征,稻瘟病菌导致稻瘟病,而赤霉镰刀菌则是小麦赤霉病的主要致病因子。全球农业贸易的加速发展进一步促进了受感染的植物材料、种子与土壤的传播,加剧了这些病原体的跨地区扩散。植物、动物与人类健康的交叉风险凸显了"同一健康(One Health)"理念的重要性,该理念强调三者间的系统互联性。无论是应对如COVID-19这类人畜共患病、口蹄疫等畜牧病,还是植物病原体如P. infestans,跨学科的综合防控措施都是维护全球健康、粮食安全与生态韧性的关键。
在全球化与国际贸易不断加速的背景下,害虫与病原体的跨境传播进一步加剧,导致新的宿主--病原体互作关系出现,从而威胁农业生产、生物多样性及生态系统稳定性。要有效缓解这些风险,建立高效而精准的诊断体系至关重要。然而,传统的人类、动物及植物病原体检测手段,如形态学观察、微生物分离培养、生化鉴定及免疫学检测等,通常耗时较长、检测灵敏度较低且依赖专业技术人员。分子生物学方法如实时定量PCR虽然可实现快速的病原体及耐药基因检测,但其检测范围有限,尤其在复杂混合感染或罕见病原体的识别中存在显著局限。
相较之下,基于测序的检测技术(如针对全基因组或特定核苷酸序列的高通量测序)因其快速、灵敏的特性而日益受到关注(图1A)。在实际应用中,全基因组测序(Whole-Genome Sequencing, WGS)结合生物信息学分析,已被广泛应用于Salmonella等病原体及其抗生素耐药基因的监测。这类技术能够在物种水平上实现微生物群落的精细解析,并在检测范围上具有显著优势。培养组学、宏基因组学、宏转录组学及靶向测序等方法,均可实现对微生物基因组的深度解析(图1B、C、D、E)。这些技术的应用范围不仅限于人类健康领域,在兽医诊断及农业防控中亦具有重要潜力,满足了发展高效、环境友好型诊断技术的迫切需求。
与以往多聚焦于单一技术或局部应用的研究相比,本文的综述提供了更为系统与综合的视角,整合了多种前沿测序技术在全球传染病监测中的最新进展与交叉应用。同时,本文强调这些技术在推动病原体检测范式转变方面的协同作用,为构建可持续的"同一健康"体系提供了重要支撑。
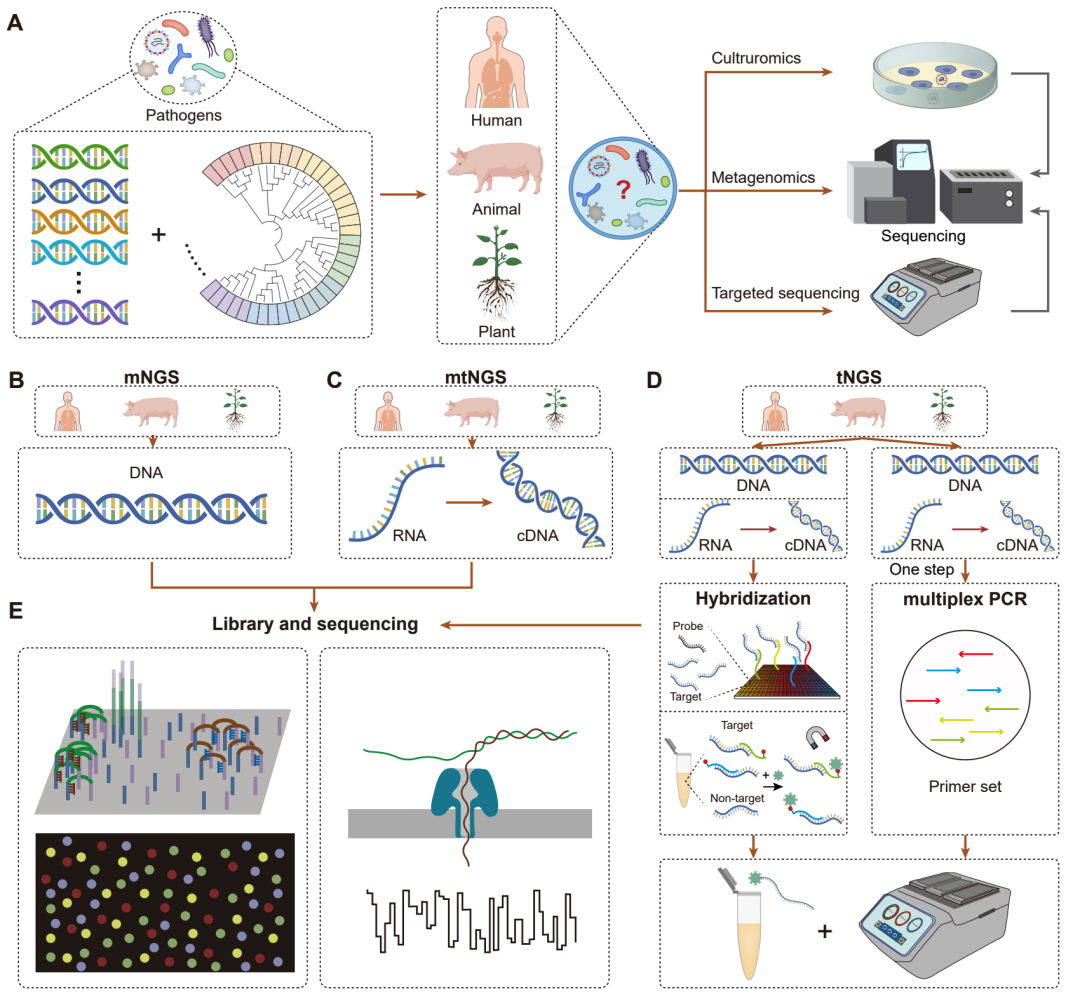
图1. 基于高通量测序的病原体检测技术
A:用于分子水平表征病原感染的高通量测序技术。B:用于病原体检测的宏基因组下一代测序(mNGS)技术。C:用于病原体检测的宏转录组下一代测序(mtNGS)技术。D:用于病原体检测的靶向测序技术。E:文库构建与测序流程。
通过培养组学探索新型病原体
在人体结肠黏膜组织及粪便样本中,约62%的未知细菌,且其中约80%的物种曾被认为无法培养。为探索这些新发现的细菌及物种,研究人员发展了多种创新方法。培养组学通过设置多样化的培养条件,实现微生物的分离与培养,并结合基质辅助激光解吸/电离飞行时间质谱(MALDI--TOF MS)或16S rRNA基因测序进行细菌鉴定,同时利用全基因组测序对物种进行重测序,从而实现精确识别。
例如,丁酸梭菌在传统宏基因组方法中无法检测其毒素分泌,但通过培养组学成功分离,并确认其与早产儿坏死性小肠结肠炎相关。这一方法对于揭示人体微生物组特征及延伸至其他研究领域具有重要价值。在动物研究中,培养组学已应用于牛、猪等畜牧动物肠道微生物组研究,揭示微生物多样性及其在健康和疾病中的作用,为提高畜牧生产力及控制人畜共患病提供了策略支持。
在植物领域,培养组学主要聚焦于根际、叶际及内生微生物,这些微生物对植物生长、抗病能力及养分循环具有重要作用。通过培养组学,研究者已成功分离出可保护作物免受真菌病害侵袭的有益微生物,从而减少对化学农药的依赖。值得注意的是,某些最初被归类为有益微生物的细菌,在临床样本中被重新评估,显示潜在致病性。例如,粘液菌属与克氏菌属虽然广泛被认为具有益生作用,但在部分临床样本中显示出病原潜力。这提示某些细菌在健康与疾病中可能发挥双重作用,亟需进一步研究以明确其潜在致病性。
然而,培养组学仍面临挑战。首先,特定微生物的生长依赖特定营养物质或环境条件,例如严格厌氧菌无法在有氧条件下生长。其次,微生物的培养与分离过程耗时较长,难以快速获得鉴定结果。因此,在紧急情况下,培养组学可能并非最优方案。
通过宏基因组和宏转录组方法进行病原体检测
宏基因组和宏转录组测序技术直接对样本中的全部核苷酸进行测序,实现宿主与微生物信息的全面获取。与培养组学不同,这些方法能够广泛识别已知及未知病原体,并发现新型微生物。宏基因组测序作为一种新兴技术,正在改变复杂临床感染的诊断方式。该方法通过对患者样本中所有微生物及宿主的DNA和RNA进行测序,提供了一种无偏倚的病原体检测手段,包括细菌、病毒、真菌及寄生虫。
在临床应用中,宏基因组测序已用于识别新生儿肺炎、败血症及脑膜炎相关的致命病原体,诊断准确率高达90%。在由李斯特菌引起的食源性暴发事件中,宏基因组测序能够有效识别病原体,并通过比较患者样本与食品样本的基因组追踪污染源。此外,该技术在免疫功能低下患者侵袭性真菌感染的诊断中也显示出价值,可直接从血液或组织中快速检测曲霉菌及多药耐药酵母,从而实现及时精准的治疗。
利用高通量宏转录组学进行的大规模病毒组研究,对中国五个哺乳动物目类的1,941只野生动物进行分析,鉴定出102种感染哺乳动物的病毒,其中65种为13个病毒科的新型病毒。该方法有效揭示了已知与未知病原体,并识别潜在的人畜共患风险,显示出宏转录组在病原体发现和监测中的重要性。
此外,宏基因组和宏转录组技术在抗生素耐药基因监测中也日益重要。宏基因组可以识别耐药基因的潜在存在,而宏转录组则可评估其实际表达情况,从而全面了解耐药基因组(resistome)的状态及传播风险。例如,对在抑病土壤中生长的甜菜根系进行宏基因组测序,可鉴定与疾病抑制相关的微生物群落及功能特征,并揭示感染过程中上调的生物合成基因簇,以重建内生微生物组。
然而,宏基因组方法也存在局限性。样本中宿主核酸比例过高是主要挑战之一。在人类样本中,宿主DNA通常超过微生物核酸的99%,导致宏基因组测序对宿主DNA丰富样本的灵敏度降低,同时增加测序深度和误差率。对于植物内生菌研究,宿主DNA同样会掩盖微生物信号,降低病原体检测的整体灵敏度。这种宿主序列的优势地位限制了低丰度微生物的检测。尽管宿主去除方法能够减少宿主污染的影响,但也可能误删低丰度微生物序列。此外,为获得完整微生物组覆盖,全基因组测序需要极高的测序深度,从而增加检测时间和成本。
通过靶向测序进行微生物鉴定与监测
全基因组测序因成本高昂和数据分析复杂,尚未在微生物鉴定中得到广泛应用。相比之下,靶向测序方法,如16S rRNA基因扩增及测序,已经成为一种成本低、速度快的替代策略,能够针对特定基因或区域进行微生物鉴定。与其他测序方法相比,靶向测序速度明显更快,因为其无需进行宿主去除实验,也不需要对整个基因组进行重测序。
靶向测序主要有两种策略:一是基于杂交的靶向测序,通过特异性探针与目标基因片段互补结合来选择性捕获和测序;二是基于多重PCR(multiplex PCR)的靶向测序,通过多重PCR富集目标基因片段再进行测序。与基于杂交的靶向测序相比,多重PCR靶向测序仅需一到两轮PCR即可完成文库构建和测序,无需芯片制造或杂交流程。这不仅降低了成本,也降低了技术门槛,使其在现场检测和即时诊断中具有显著应用潜力。
然而,靶向测序的准确性高度依赖引物或探针的选择。截至目前,16S rRNA检测中已使用超过500对引物,研究表明,不当的引物选择可能导致对某些物种的代表性不足或偏倚。因此,引物的设计与选择是靶向测序中的关键挑战。
在靶向测序方法中,多重PCR靶向测序尤为引人注目,它能够同时富集并测序多个目标,适合低丰度及混合样本的高性价比检测。研究表明,在呼吸道病原体检测中,多重PCR靶向测序的成本仅为宏基因组测序的四分之一。在COVID-19大流行期间,该技术被广泛用于分析人类及污水样本中SARS-CoV-2的谱系变异、监测病毒丰度、追踪感染源及评估个体治疗反应。其应用随后扩展至其他与人类相关的病原体。此外,作为多重PCR靶向测序的一部分,扩增子测序(amplicon sequencing)已成为环境及宿主系统中微生物群落特征研究的几乎通用工具。基于16S和ITS的扩增子测序方法,已被用于评估入侵植物白花蛇舌草叶片上的致病真菌与共生本地植物的相似性,也用于甘蓝土壤病原菌以及辣椒根、茎、叶、果等12个微生物组的细菌和真菌群落研究。这一技术显著拓展了环境DNA微生物组的认识。
尽管多重PCR靶向测序具有成本效益和广泛应用,但在准确区分物种水平差异及均衡识别所有目标方面仍面临挑战。这主要源于微生物的多样性和丰度差异,以及引物选择对特定细菌类群代表性的显著影响。多重PCR靶向测序的准确性高度依赖引物设计。此外,随着引物数量的增加,引物二聚体形成和非特异性扩增问题加剧,导致检测范围与多引物兼容性之间存在冲突。
多重PCR靶向测序的挑战与策略
多重PCR靶向测序因其操作简便、富集效率高,已被科研人员和商业机构广泛采用。这一方法在检测低频肿瘤体细胞突变以及分析保守序列中丰富变异以识别病原体方面表现尤为出色。引物在PCR靶向测序中起着核心作用,其设计直接影响检测能力。然而,由于目标序列高度多样化,设计能够检测传染性流行病病原体的通用PCR引物(如针对细菌16S rDNA区域的引物)一直具有挑战性。
除了细菌之外,样本中可能存在的病毒、真菌、支原体、衣原体及寄生虫等其他感染因子,也增加了靶向测序引物设计的复杂性。常用的策略是针对每种病原体类型单独设计引物,然后组合使用,但这种方法增加了引物二聚体形成及非特异性扩增的风险,可能导致PCR失败。因此,引物优化设计仍然是多重PCR靶向测序的关键难点。
目前,多重PCR系统的引物设计主要采用三种策略,并配合不同的软件工具。第一种策略是对每条目标序列分别设计引物(如使用Primer3软件),然后进行整合。候选引物需经过两轮质量控制:初轮QC1利用在线BLAST工具检测潜在的非特异性问题,并评估引物二级结构以提升扩增效率;次轮QC2则全面检测候选引物间的兼容性及特异性,以消除引物二聚体和非特异性扩增。然而,该策略耗时且劳动强度大,不适合高通量应用(图2A)。
第二种策略基于K-mer生成引物,通过将输入序列切割成短K-mer,并选择高频K-mer设计高覆盖率引物,再组装成引物集合(图2B)。这种方法在保证目标覆盖率的同时,减少了引物兼容性和特异性评估的工作量。第一轮质量控制评估参数包括目标和宿主基因组中的GC含量及K-mer频率。
第三种策略利用多序列比对识别保守区域,以设计简并引物(degenerate primers)(图2C)。质量控制与第二种策略类似。简并引物能够提供更广泛的目标覆盖,并简化设计流程,主要聚焦于保守区域。尽管可将简并引物与K-mer策略结合以扩展覆盖率,但高多样性的K-mer需要额外的计算资源和时间才能有效整合简并碱基。总体而言,虽然多种方法已用于引物设计,但大部分不使用简并引物,应用范围有限。
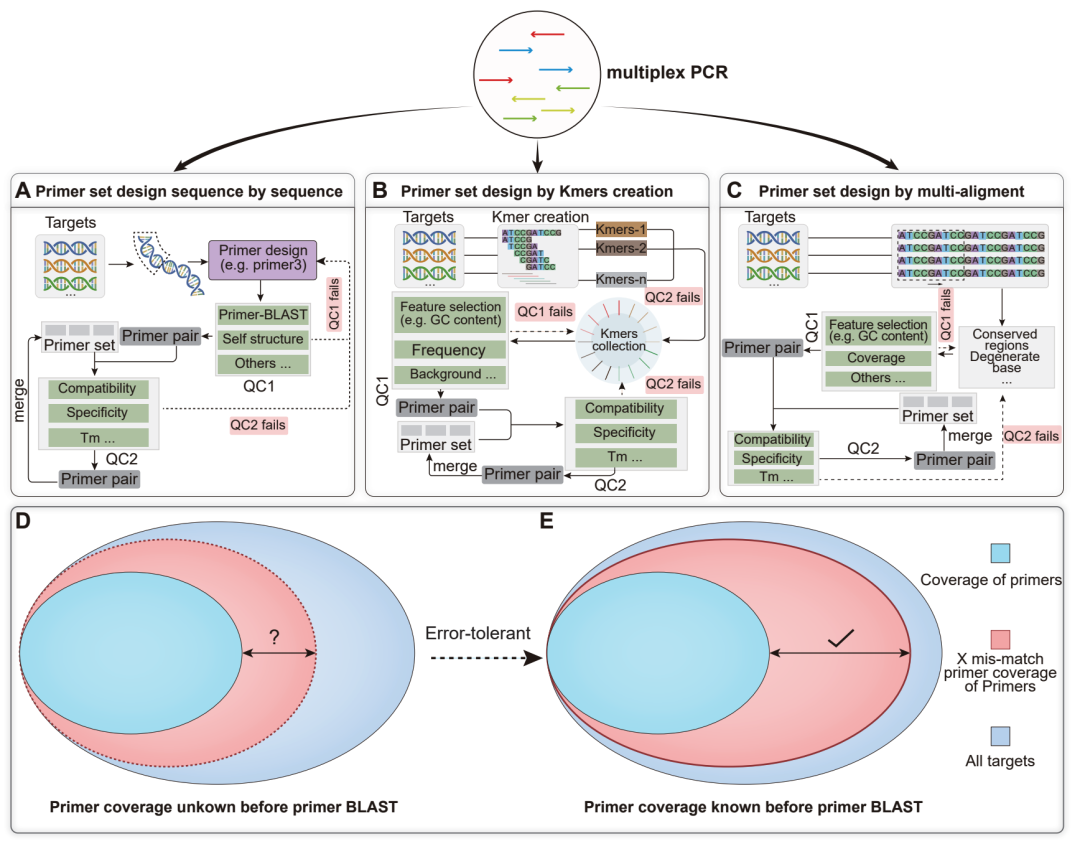
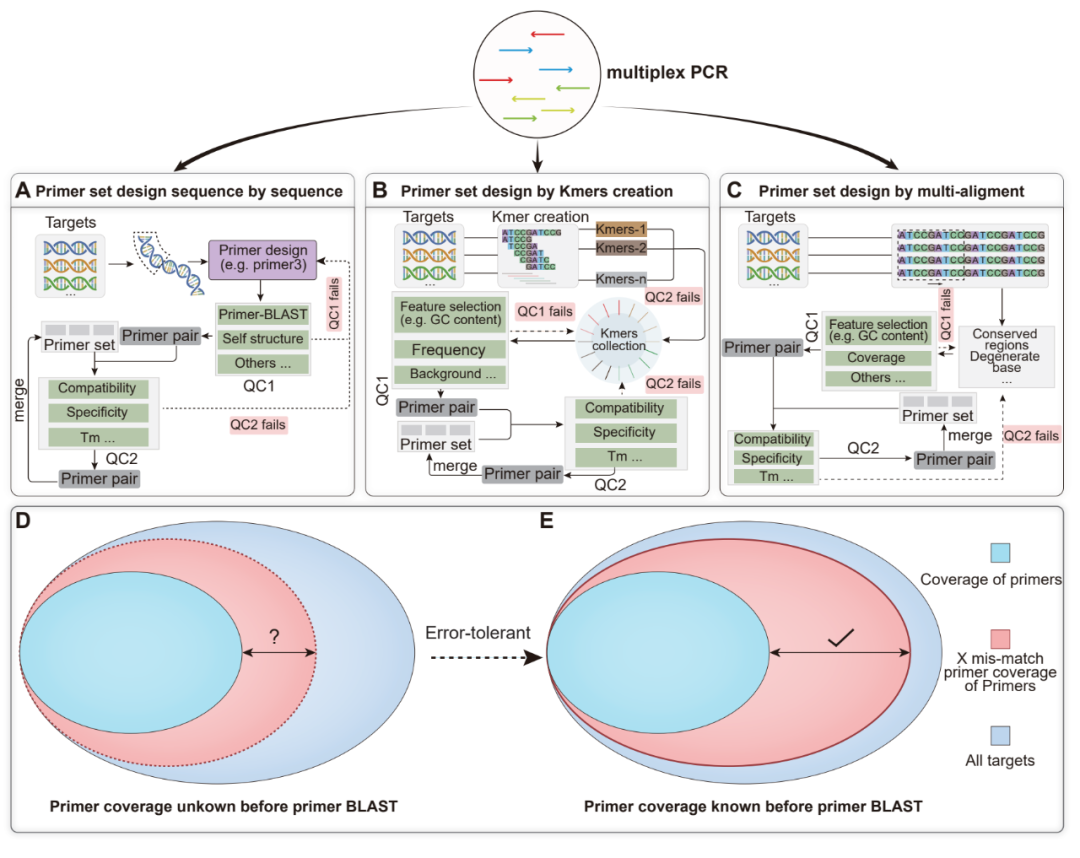
图 2. 误差容忍型简并引物的优势
A:逐条序列设计的引物集合。B:通过k-mer生成的引物集合设计。C:通过多序列比对的引物集合设计。D:采用完全匹配模式的引物设计。E:采用误差容忍模式的引物设计。QC:质量控制
容错策略:提高PCR靶向测序效率
通常,引物设计旨在最大程度减少碱基错误配对,同时保证覆盖率、特异性和灵敏度。然而,研究表明,当匹配碱基的热力学稳定性超过不匹配碱基时,即使引物含有错配碱基,也仍可能启动PCR扩增。因此,为实现完整覆盖,可以使用包含错配碱基的引物。
现有大多数引物设计方法仅基于引物与模板的完全匹配来生成最大覆盖率的引物工具,因此无法在允许一定数量错配(X错误,X可为0、1或2)(图2D)情况下实现最大覆盖。为此,可在引物设计中权衡错配数量与引物特异性和灵敏度,以获得更高覆盖率的容错引物(图2E)。
理论上,一组512条引物即可覆盖10个目标(图3A),但实际情况极少出现。原因在于512条引物同时存在会显著增加引物二聚体形成和非特异性扩增的风险,同时每条引物的最终浓度会比正常PCR低512倍,从而影响引物与模板的退火效率,降低PCR效率。如果引物简并度不超过4,则完全匹配时最大覆盖率仅能覆盖10个目标中的4个(图3B)。实现完整覆盖通常需要至少四轮引物设计,每轮选择覆盖率最高的引物,这不仅增加了引物数量,也使设计过程更复杂,因为每轮都需考虑新设计引物与现有引物之间的兼容性。
采用容错模式时,如果允许引物与目标序列之间存在最多一个错误,两条简并度为4的候选引物即可实现100%覆盖率(图3B)。由此,每条引物的最终浓度仅为初始浓度的1/4。有限简并度的引物设计可确保所有引物的最终浓度相对较高。需要注意的是,容错模式设计的简并引物中简并碱基的组成,与完全匹配最大覆盖设计的引物不同(图3B)。此外,还可通过优化简并碱基及错配位置进一步提高性能。例如,引物二聚体主要通过3′端互补配对形成,因此将简并碱基远离引物3′端可降低二聚体形成的概率。同时,3′端的错配可能显著影响引物与模板的结合效率,因此应尽量避免3′端碱基错配,以维持引物的特异性和灵敏度。
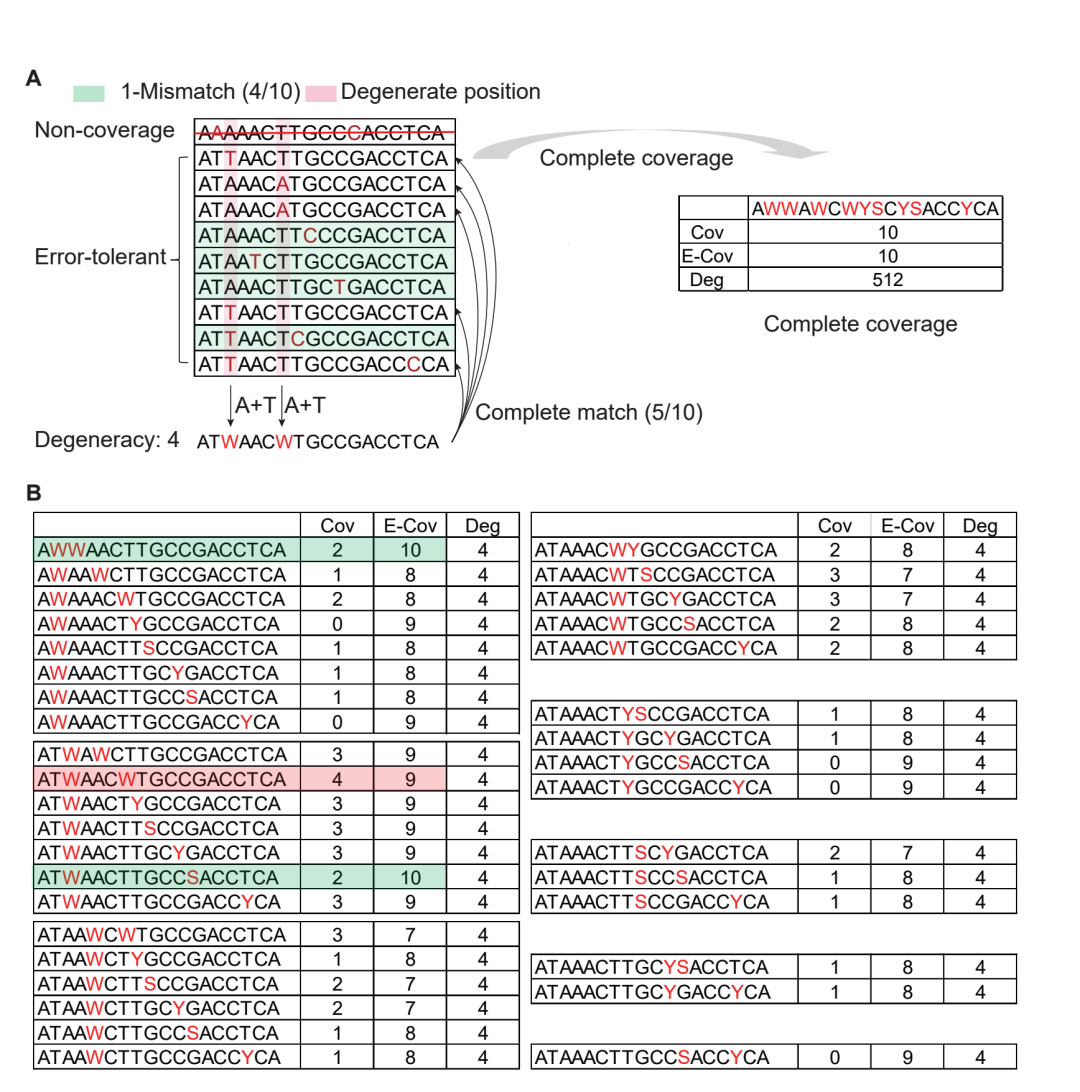
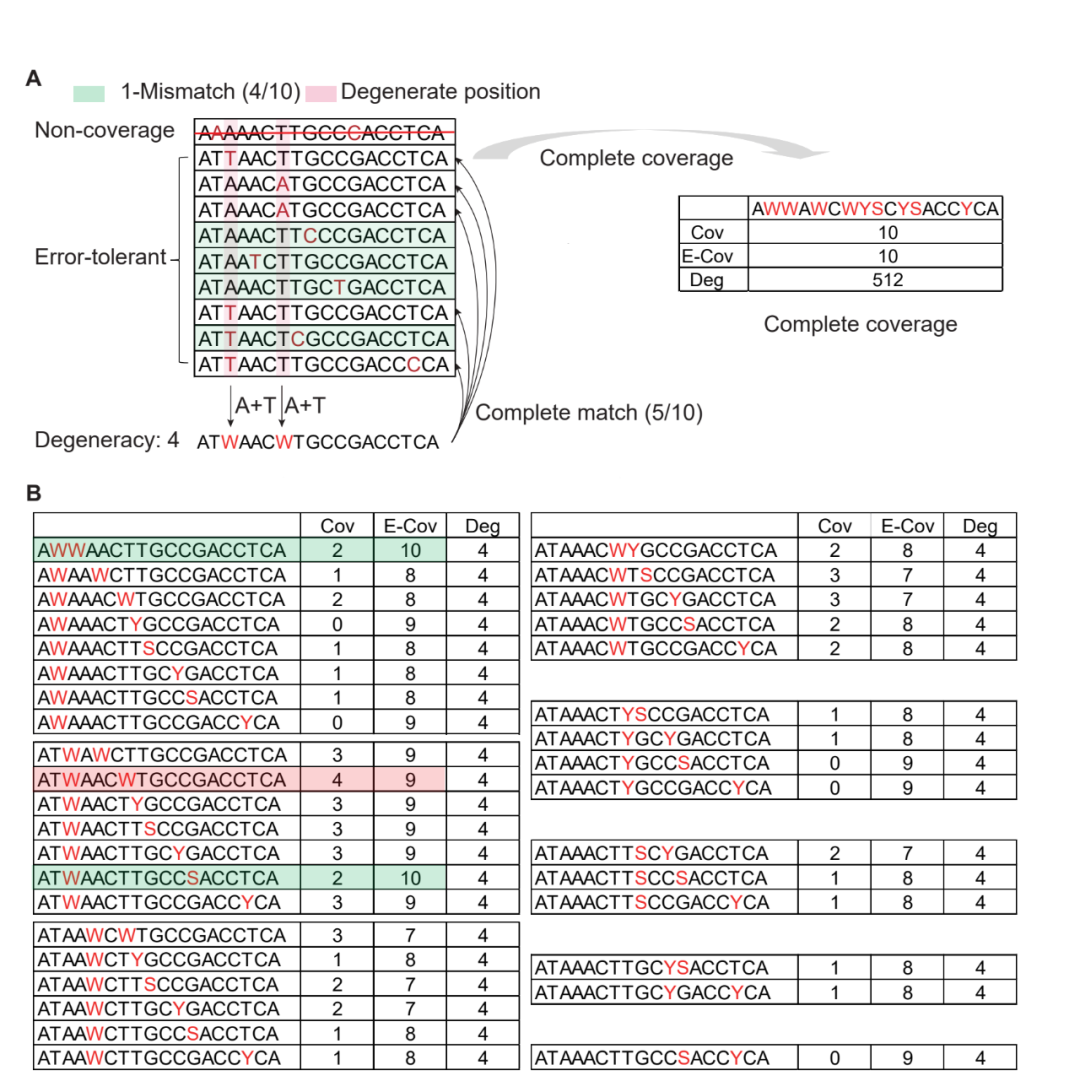
图 3. 多重PCR引物设计中的覆盖度与误差容忍模型
A:多重PCR目标区域的示意图。"完全匹配(Complete Match)":指与模板序列完全匹配的引物;"完全覆盖(Complete Coverage)":指在完全匹配模型中能够结合所有输入序列的简并引物;"单碱基错配(1-Mismatch)":指与模板序列存在一个碱基错配的引物。B:完全覆盖设计方法与误差容忍模型的示意图。红色标注的简并引物代表具有最大完全覆盖能力的引物,绿色标注的简并引物代表具有最大误差容忍覆盖能力的引物。Cov:具有完全覆盖的引物;E-Cov:具有误差容忍覆盖的引物;Deg:简并度。
容错简并引物设计的挑战
容错引物的设计属于经典的非确定性多项式(NP)完全问题。为设计简并引物,研究者已经开发了多种程序。一个经典的容错简并引物设计程序是 HYDEN,它接受多序列比对格式的DNA序列作为输入,并生成覆盖所有输入序列的候选引物集合。相比之下,Primux 不依赖多序列比对,而是采用基于K-mer的策略进行引物设计。然而,这两类程序均计算量大,耗时较长,这主要源于序列多样性高及K-mer数量庞大。两者都对碱基错配位置敏感,但缺乏控制错配位置和识别错配类型的模块。
相比之下,MultiPrime 在处理这些问题上表现较好。它首先对输入序列进行聚类,然后从聚类中随机抽样,并根据用户需求设计引物,例如设定简并度、简并碱基位置、允许的错误数量及错配位置。然而,该程序仍无法全面评估容错引物的效率和特异性。总体而言,针对引物容错性质的优化实验相对较少,尚未确定设计最大容错覆盖率引物的最佳方法。因此,精确评估碱基错配数量、位置及类型对引物效率和特异性的影响,对于实现多重PCR靶向测序的实用化至关重要。
人工智能在测序技术中的应用:革新病原检测与引物设计
人工智能(AI)将在组学技术和靶向测序技术的发展中继续发挥重要作用,对病原微生物的检测、预防与治疗产生深远影响。AI与测序技术的结合正在变革人类、动物和植物病原体的检测,使微生物病原体的识别更加快速、精准且可扩展(图4)。
在人类病原体检测中,AI通过克服宿主主导的核酸信号,提升了低丰度病原体的诊断灵敏度,并改善了多重微生物感染的识别。例如,将宏基因组测序与AI算法结合,可从复杂临床样本中提取微生物特征,对于新兴传染病原体及抗菌耐药菌的检测尤为关键。此外,机器学习(ML)已被有效应用于感染生物学相关显微镜数据的处理。各种显微技术,包括光学显微镜和电子显微镜,均已促进ML模型的发展,使其能够在宿主细胞中检测细菌、真菌、寄生虫和病毒。
在兽医诊断领域,AI增强的测序工具已用于应对重大牲畜病原体,如非洲猪瘟病毒、禽流感病毒和口蹄疫病毒,实现快速病原识别,从而预防疫情爆发。此外,AI优化的纳米孔测序已应用于食品源性人畜共患病原体的检测,具有高灵敏度和高特异性。测序技术在兽医实践中的整合,也有助于监测牲畜微生物耐药基因。
在农业领域,AI驱动的测序技术增强了植物病原体(包括细菌、真菌和病毒)的识别能力,例如通过16S rDNA测序。这些技术利用AI优化引物设计,提高低丰度微生物群体的检测特异性,如在观赏植物中的应用所示。AI还被用于预测冬大麦网斑病的发展,为病害诊断提供了新的方法,具有高诊断准确性、预测精度和田间适用性。这类创新有助于作物病害的早期发现,减少对化学农药的依赖,从而促进环境可持续的农业实践。
在各个领域中,AI驱动的测序技术带来了诸多创新,例如容错引物设计、宿主DNA去除以提高灵敏度,以及可实现现场实时诊断的便携式测序平台,使在资源有限或田间环境中开展检测成为可能。AI与测序技术的协同作用正在改变人类、兽医和农业领域的微生物监测及疾病管理模式。
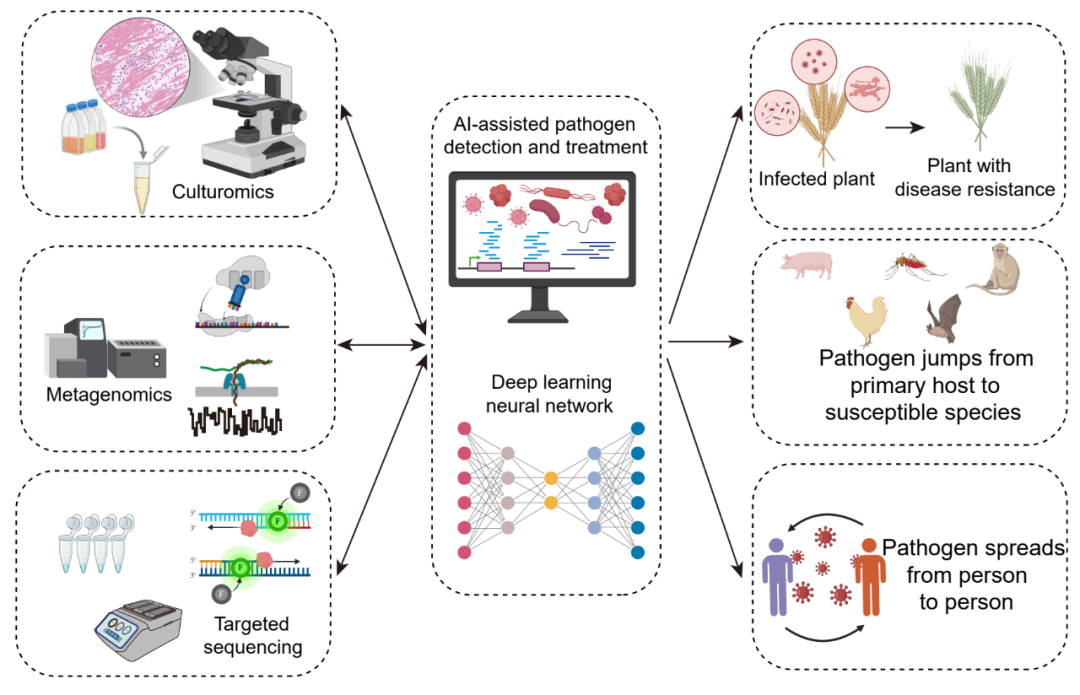
图 4. 人工智能在促进培养组学、组学技术及靶向测序技术发展中的作用
结论
应对传染性疾病的挑战需要多层次的方法,整合先进的分子技术与对病原多样性及诊断难题的深入理解。高通量测序,尤其是基于多重PCR的靶向测序,已显示出其在病原检测中的潜力,能够提供快速、精准且成本效益高的解决方案。其同时富集并测序多个靶标的能力,使其成为应对异质样本及多样化病原体复杂性的不可或缺工具。然而,没有单一技术能够成为通用方案。例如,培养组学通过提供高质量分离株实现高准确性,但无法检测不可培养物种;宏基因组学能够提供微生物群落的全面调查,但计算复杂且成本高;转录组学可揭示微生物的活跃状态,但成本和操作要求更高;靶向测序虽提供低成本、高灵敏度的解决方案,但难以发现新型病原体。因此,针对具体诊断需求和研究目标,合理选择并整合这些策略至关重要。
针对实际应用的具体需求,我们提出如下建议:对于已知病原体的检测,应采用靶向策略:可从NCBI RefSeq获取参考基因组,通过MultiPrime设计特定基因引物,并使用Bowtie 2等比对工具及SAMtools、BCFtools等变异分析工具处理数据。相反,对于新型病原体的检测,需要采用无参考的宏基因组方法,可使用基于K-mer的分类器(如Kraken 2)进行快速鉴定,使用MEGAHIT和SPAdes等组装软件进行基因组重建,并通过EasyMetagenome等综合性分析流程完成发现。病原体检测策略还应针对宿主类型进行调整。在动物病原体检测中,应通过Primer-BLAST等工具确认引物特异性,以避免宿主交叉反应。在植物病原体检测中,由于宿主DNA污染和病原体多样性是主要挑战,可借助VEuPathDB等资源,并通过专用分析流程如PhytoPipe处理RNA-seq数据以提高分析效率。
尽管基于测序的病原体检测前景广阔,但仍需解决若干关键问题以确保其可靠性和广泛应用。主要挑战之一是生物信息数据的管理与解读,特别是分析工具性能差异引入的假阳性和不一致性风险。鉴于误诊的潜在高成本,未来工作必须优先开发稳健、标准化的分析框架,结合严格的质控、共识算法方法和正交验证,确保数据可靠性。此外,由于NGS可发现新型微生物,在从计算预测到功能验证的过程中建立明确的风险评估流程,对全球生物安全至关重要。
展望未来,先进计算工具,尤其是AI的整合,前景广阔。除了提升分析速度和可扩展性外,下一个前沿方向是可解释AI(XAI)的开发,通过结合生物网络等领域知识,提高模型的可解释性。这对于优化容错引物设计、提升检测准确性以及理解复杂宏基因组数据至关重要。
当这些当前限制通过创新和协作研究得到解决时,基于测序的技术将在临床、兽医和农业领域得到广泛应用。这些进展不仅有助于提升诊断水平,还将推动全球健康韧性、可持续农业实践和稳健的疾病管理框架的发展。持续探索和优化这些技术,同时强调数据完整性和负责任的创新,对于在日益互联的世界中保障健康和生物多样性至关重要。
作者简介
第一作者简介
罗豪

罗豪,中国农业科学院深圳农业基因组研究所,在读博士研究生;研究方向为微生物组方法开发与应用,在iMeta、Advanced Science、iMetaOmics、GigaScience、aBIOTECH等期刊发表论文8篇,其中第一/共同第一作者5篇。
王瑶

王瑶,农学博士,毕业于德国哥廷根大学,主要研究方向为植物病理、微生物组、植物-微生物互作。近5年,以第一/共通讯在iMeta、PLoS Pathogens等期刊发表十余篇文章。获德国植物病理协会青年科学家奖、广东省海外人才支持项目等。
通讯作者简介
杨军波

杨军波,副研究员。中科院遗传所博士,北京大学博士后,中国农业科学院深圳农业基因组研究所博士后。目前研究的主要课题为遗传学,微生物组学,表观遗传学和生物信息学等方向。
刘永鑫

刘永鑫,中国农科院深圳基因组所研究员,iMeta执行主编。聚焦微生物组方法开发、功能挖掘和科学传播,在Nature Biotechnology、Nature Microbiology等发表论文80篇,14篇入选ESI高被引论文,被引29000+次,入选全球前2%科学家榜单、国家级青年人才项目。兼任中国微生物组、计算合成生物学专委会委员,创办18万+同行关注的宏基因组公众号,主编《微生物组实验手册》专著,发起iMeta 期刊(IF 33.2),位列全球前千分之三。兼职为Cell子刊、NC、NAR、Microbiome等期刊审稿360余次。
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
iMeta高引 fastp PhyloSuite ImageGP2iNAP2 ggClusterNet2
iMeta工具 SangerBox2 美吉2024 OmicStudioWekemo OmicShare
iMeta综述 高脂饮食菌群 发酵中药 口腔菌群 微塑料 癌症 宿主代谢
10000+:扩增子EasyAmplicon 比较基因组JCVI 序列分析SeqKit2 维恩图EVenn