
研究论文
● 期刊:Cell(IF:42.5)
● DOI:https://doi.org/10.1016/j.cell.2025.08.020
●原文链接: https://www.cell.com/cell/fulltext/S0092-8674(25)00975-4
● 第一作者:Jeremiah J. Minich
● 通讯作者:Jeremiah J. Minich (jeremiah.minich@gmail.com), Rob Knight (robknight@ucsd.edu), Mark J. Manary (manarymj@wustl.edu), Todd P. Michael (tmichael@salk.edu)
● 发表日期:2025-09-09
● 主要单位:
植物分子与细胞生物学实验室,萨克生物研究所,美国加利福尼亚州拉霍亚(The Plant Molecular and Cellular Biology Laboratory, The Salk Institute for Biological Studies, La Jolla)
加利福尼亚大学圣地亚哥分校儿科系,美国加利福尼亚州圣地亚哥(The Plant Molecular and Cellular Biology Laboratory, The Salk Institute for Biological Studies, La Jolla)
华盛顿大学圣路易斯分校儿科系,美国密苏里州圣路易斯(Department of Pediatrics, Washington University in St. Louis, St. Louis)
摘要Abstract
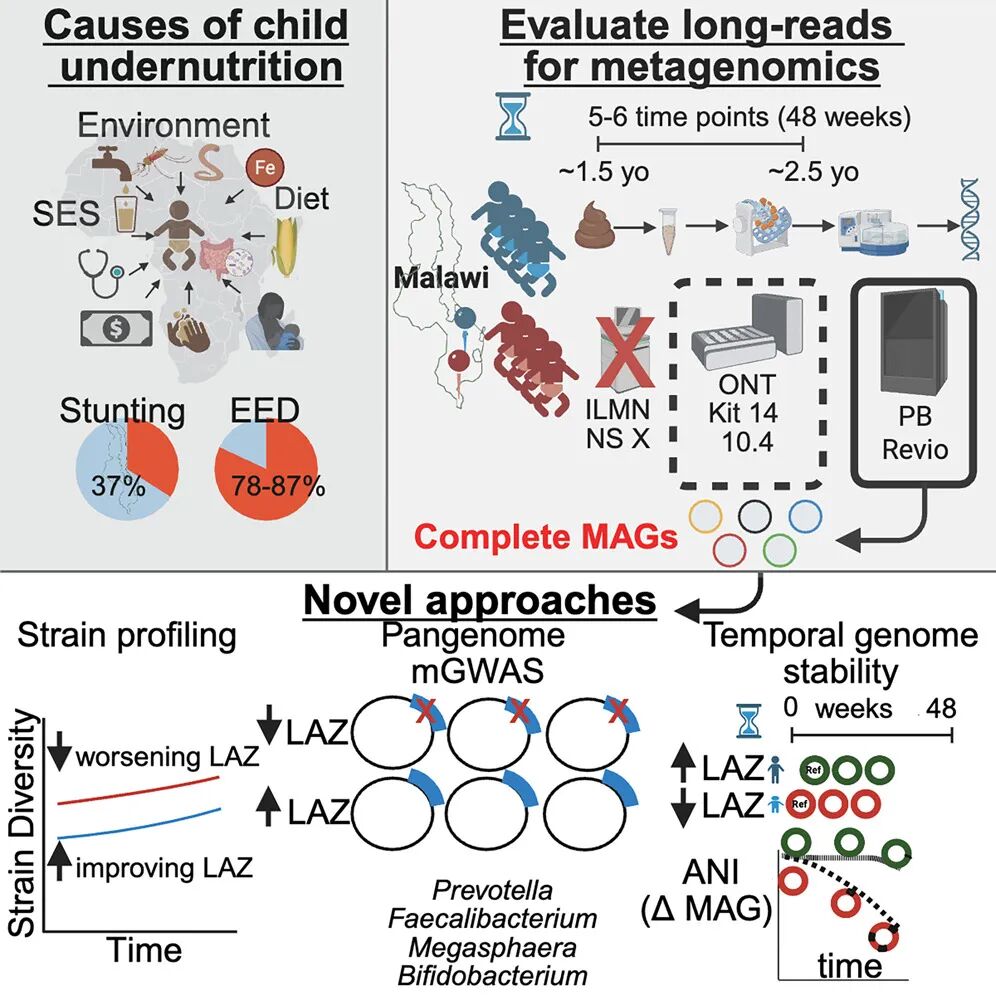
人类肠道微生物组与儿童营养不良相关,但传统的微生物组研究方法缺乏足够的分辨率。我们假设,通过长读(LR)DNA测序恢复的完整宏基因组组装基因组(cMAGs)能够促进泛基因组和微生物全基因组关联研究(GWAS),以识别与儿童线性生长相关的微生物基因关联。与短读方法相比,长读方法每千兆碱基对(Gbp)产生的cMAGs数量是短读方法的44-64倍,其中PacBio(PB)提供了最准确且具有成本效益的组装结果。在马拉维的一个纵向儿科队列中,我们从47个样本中生成了986个cMAGs(其中839个为圆形的),并将该数据库应用于扩展的210个样本集。通过机器学习方法,我们识别出了与线性生长相关的物种。泛基因组分析揭示了与线性生长相关的微生物基因关联,而基因组不稳定性与年龄-身高Z评分(LAZ)的下降相关。该资源展示了将cMAGs与健康轨迹进行比较的强大能力,并为微生物组关联研究确立了新的标准。
引言Introduction
全球范围内,约有1.492亿名5岁以下儿童(22%)患有营养不良(身高低于年龄标准),其中有4540万儿童(6.7%)患有消瘦(体重低于身高标准)。中度急性营养不良(MAM),其定义为体重与身长的Z值(WLZ)<−2,或上臂围为115--124毫米;而重度急性营养不良(SAM),其定义为WLZ <−3,或者孩子出现双侧凹陷性水肿,或上臂围<115毫米,通常影响1--5岁的儿童。这些是更为严重的营养不良形式,但其发生率较低。生长迟缓(stunting),即身长与年龄的Z值(LAZ)<−2,是最常见的营养不良形式。生命最初1000天的营养不良尤其有害,可能导致不可逆的长期认知和发育损伤,影响学业成绩、经济状况,并对母体健康产生负面影响。东南亚和撒哈拉以南非洲(SSA)的营养不良水平最高,马拉维的生长迟缓发生率高达37%。
通常,线性生长的变化(ΔLAZ)在几周的时间框架内进行测量,并通过(当前时间点的LAZ - 之前时间点的LAZ)来计算。从某一时间点得到的负值表示LAZ在下降,而正值则表示LAZ在上升。一种假设的线性生长障碍原因是长期炎症性、功能失调的肠道,通常由微生物群失调引起,导致营养吸收受损。环境肠道功能障碍(EED)是一种小肠的临床亚急性、慢性炎症性疾病,特征为绒毛萎缩、粘膜T细胞浸润和肠道通透性增加。EED与儿童的线性生长受限有着持续的关联。尽管对消瘦和生长迟缓进行了数十年的研究,但其潜在机制和广泛有效的干预措施仍未明确。大多数研究集中在静态终点,如体重与身高的Z值(WHZ)、LAZ或单一时间点的EED。然而,动态指标,如LAZ的变化率(ΔLAZ),反映了线性生长速度,可能为儿童健康提供更为细致的见解。这些纵向标记尚未得到充分探索,但却至关重要。我们的研究旨在填补这一空白,专注于通过LAZ的变化来衡量线性生长。
Smith 等人首次证明了微生物组、饮食与马拉维双胞胎中重度急性营养不良(SAM)之间的因果关系。从那时起,研究已将肠道微生物群失调和微生物组未成熟与营养不良联系起来。2024年,Chibuye 等人的综述回顾了14项关于微生物组与生长迟缓的研究,但其中只有两项使用了全基因组宏基因组学(shotgun metagenomics);其余的则依赖于16S rRNA。尽管样本量较小(马拉维队列18人;孟加拉队列11人),且联合设计使解释更为复杂,两个宏基因组学研究显示生长迟缓儿童的多样性降低。Surono 等人使用较大的印度尼西亚队列(78名生长迟缓儿童,53名非生长迟缓儿童)报告称生长迟缓儿童的多样性略高,但仅评估了重度生长迟缓(LAZ <−3)。14项研究中只有5项共享了测序数据,这限制了二次分析,突显了可获取基因组资源的迫切需求。营养不良微生物组的研究通常侧重于识别与健康结果相关的分类群相对丰度变化,较少关注基因功能,通常基于单一时间点的"横断面设计"。然而,纵向采样已改善了对肠道疾病的预测,即使仅使用16S数据。通过比较基因组学、泛基因组学、微生物GWAS等方法,在时间框架内使用微生物基因组识别重要分类群中的基因关联,解决因果关系与相关性之间的差异,显然存在一个明确的机会。
宏基因组学的目标是从混合群落中解析完整的微生物基因组,并在不同样本之间进行代谢能力的分析。微生物基因组组装(metagenome-assembled genomes,MAGs)首次在2004年对酸性矿山排水样本中进行。过去20年中,借助下一代测序技术的进展,MAGs已从地球上几乎每个环境中成功恢复。尽管这些短读(SR)测序技术每个研究能够获得数万个MAGs,但这些MAGs通常是高度碎片化且不完整的。获取完整的基因组能够捕获跨多个样本的完整遗传信息,从而通过泛基因组分析和微生物全基因组关联研究(mGWASs)等方法,稳健地识别与特定表型相关的功能基因等基因内容。微生物泛基因组分析已成为揭示病原体(培养分离物)遗传机制的有力工具。然而,使用MAGs进行的泛基因组分析具有挑战性,因为这些基因组通常是高度碎片化且不完整的,这会导致一些有争议的解释,因为大多数计算分析依赖于基因的存在或缺失的比较。
使用PacBio(PB)或Oxford nanopore Technologies(ONT)的长读(LR)单分子DNA测序有潜力从宏基因组中解析完整的微生物基因组。尽管合成长读或连接长读方法(例如TELL-seq)在宏基因组样本中已有几年表现出希望,Illumina(ILMn)于2022年推出的novaSeqX将测序成本降低至每千兆碱基对(Gbp)2美元。这些长读技术的进步催生了许多新的宏基因组学计算工具,尽管在考虑跨平台比较时,数据可重复性仍存在许多问题。长读宏基因组学的其他重大挑战包括样本处理的低通量和高成本,这导致许多研究样本量较小(例如<5个),或由简单、低多样性和模拟群落主导。尽管长读宏基因组学具有改变微生物生态学领域,包括营养研究的潜力,但由于通量、成本的限制以及跨平台数据可重复性存 在不确定性,它尚未被广泛采用。
利用长读(LR)宏基因组测序来量化菌株级的分类丰度、进行物种约束的遗传分析,并跟踪细菌进化,对于全球健康具有变革性的潜力。当应用于偏远的现场分子实验室时,这种方法能够为大流行监测、抗生素抗性、传染病诊断以及营养不良的微生物贡献提供实时洞察。我们使用ONT、PB和ILMn SLR技术,生成了一个关于儿童营养不良的纵向开放访问的LR宏基因组资源。我们为47个样本应用了高通量DNA提取管道,使用PB(591 Gbp)和ONT(1,172 Gbp)进行了测序,42个样本也使用ILMn(625 Gbp)进行了测序。此项工作产生了986个完整的cMAGs(839个为圆形),代表了363个物种,包括74个推测的新物种(ANI < 95%)。该定制基因组数据库支持跨平台微生物特征分析,并在扩展的42个参与者、五个时间点的数据集中改善了特征解析,通过机器学习揭示了线性生长的分类学和功能预测因子。我们还进行了泛基因组和微生物全基因组关联研究(mGWAS)分析,涵盖了超过10个谱系,并研究了噬菌体驱动的基因组进化随时间的变化。
结果Results
我们假设,LR宏基因组测序能够生成cMAGs,这些cMAGs将:(1)改善传统的分类学分析,提供更高分辨率,从而进行强有力的多样性分析;(2)启用一种新的物种约束分析方法,识别与元数据类别相关的菌株之间的基因关联(宏基因组分析);(3)揭示这些临床试验中的基因功能,试验对象为来自两个不同村庄的12至24个月大的儿童;(4)追踪与关键元数据类别相关的物种。图1A展示了这两个村庄在急性营养不良、身高矮小和肠道炎症性疾病(EED)方面存在显著差异。在360名较大年龄组的儿童中,我们选择了8名儿童进行微生物组分析,分析时将其按线性生长、性别和村庄分组。该队列被选为前瞻性纵向采样设计,并且包括了详细的农村撒哈拉以南非洲婴儿的体格测量数据,这些婴儿群体面临着营养不良及其长期后果的高风险。该婴儿群体的社会经济和人口统计特征反映了许多撒哈拉以南非洲农村地区的现实。我们还关注了结构变异(SV)和水平基因转移(HGT)在细菌菌株中的演化,这些菌株来自纵向采样研究中的婴儿群体。
为了本概念验证研究的范围,确定了45-50个样本作为可行且合适的目标。在从360名婴儿队列中选择,应用了几个标准。首先,要求具备纵向数据,因此任何少于五个时间点粪便采样的参与者被排除。其次,由于线性生长是主要的临床变量,并且样本量有限,因此我们选择了具有一致正向或负向线性生长轨迹的参与者,线性生长由LAZ(年龄-身长Z评分)定义。第三,努力在增长和减少LAZ结果之间平衡年龄、性别和诊所地点。
尽管有这些筛选条件,子集在年龄(中位数19.3个月 vs. 17.8个月)、性别(50% vs. 54%为男性)、上臂中部围度(中位数15.4 cm vs. 14.6 cm)和年龄-身长Z评分(−1.48 vs. −1.57)等其他变量方面,与总体队列相似(见Mendeley数据中的表S1)。所有8名参与者的营养终点变化的详细信息见Mendeley数据中的表S2)。每个孩子提供了五到六个粪便样本,样本间隔约11个月(图1B)。线性生长轨迹是一个重要的临床结局,也是我们在时间序列微生物组采样中评估的表型之一(图1C;见Mendeley数据中的表S2)。
我们开发了一种高通量、高分子量(HMW)DNA提取方法,兼容多种LR技术(图1D和1E)。我们选取了8个粪便样本(每个参与者第48周的样本)、一个阴性对照和三个不同的阳性对照,用于评估五种HMW DNA提取方法。我们选择了临床试验的最后一周进行采样,因为那时多样性可能是最高的,因此最为复杂。修改后的MagMax Microbiome试剂盒在所有试剂盒中DNA产量最高(p = 0.0002,Friedman统计量 = 21.96),每个样本的平均粪便DNA产量为6,154 ng(图S1A)。我们进一步优化了该方法,使得清理步骤能够通过KingFisherFlex上的定制"慢速"方法进行高通量操作,从而减少了DNA剪切,并使用该方法从完整数据集中提取了DNA(图1D)。
从47个粪便样本中,我们生成了88个PB连接文库、81个ONT连接文库、47个ONT快速文库和40个ILMn SLR(ILMn novaSeqX合成LR与Tell-Seq),总的设备外产量分别为591 Gbp(PB)、1,172 Gbp(ONT)和625 Gbp(ILMn)(图S1A)。我们优化了库制备(图S1B)和测序(图S1C)的DNA输入要求。测序摘要统计数据显示,每次测序的产量对于PB为39.28 Gbp(标准差±12.42),而ONT为56.85 Gbp(标准差±32.79)(p > 0.05)。PacBio在中位读长质量(QV 37.27,平均值,标准差±1.69 vs. 17.23,平均值,标准差±1.27,p < 0.0001)、平均读长(p < 0.0001)、中位读长(p < 0.0001)和n50读长(p < 0.0001)方面显著优于ONT,而ONT的最大读长更长(p < 0.0001)(图S1D--S1M)。对于LR测序而言,平均读长N50是一个关键指标,ONT的N50为8,712 bp(标准差±5,979),PB为9,663 bp(标准差±1,868),这两个值与其他已发表的研究结果高度可比(图S1L)。值得注意的是,我们的PB平均测序运行产量(Gbp)远低于其宣传的90 Gbp值。我们预计随着文库加载的优化,这些输出将会增加,并且通过几次超过50 Gbp的运行,我们观察到了改进(图S1C)。随着基础调用模型的改进和针对细菌基因组的调优,ONT的质量也预计会随着时间的推移而提高。
我们将本研究与另外30项LR宏基因组研究进行了比较,发现本研究在几个关键方面具有独特性。它是唯一一项使用并直接比较最新技术进展的研究,涉及ONT(Kit 14和R10.4.1)和PB(Revio和SMRTbell 3.0)两种平台。我们的数据集也是迄今为止两种平台中最大样本量和总测序产量的研究(图1F;见Mendeley中的表S6)。重要的是,我们的研究包括了纵向组件,使其与其他研究进一步区别开来。尽管有两项先前的研究包含了纵向采样,但它们都依赖于较旧的PB Sequel技术,并且范围有限:一项分析了海水样本(n = 4),另一项分析了奶酪样本(n = 15)。
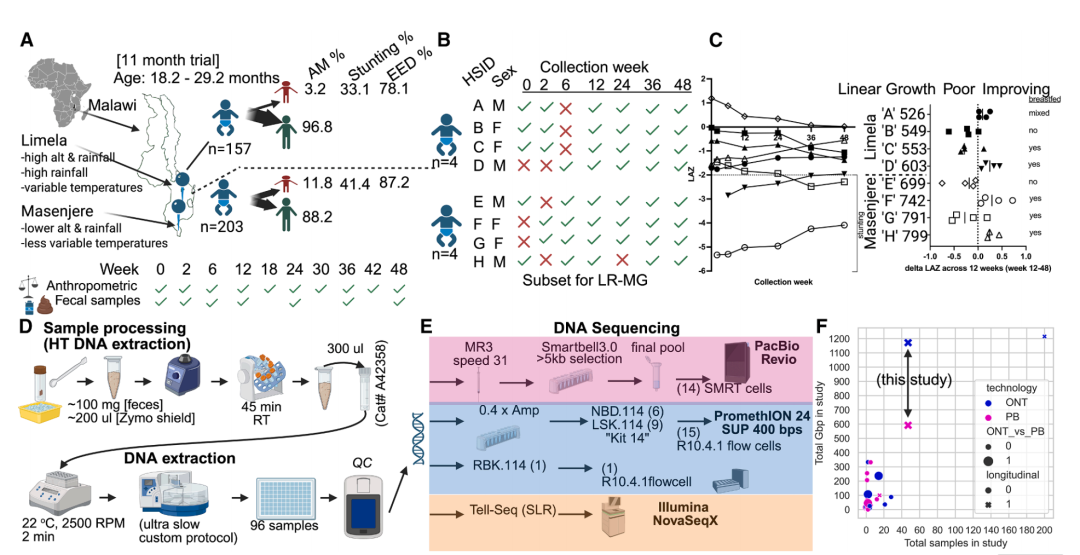
图1 | LR宏基因组评估的研究设计
(A) 原始临床试验设计,评估了两个村庄,其中婴儿在最多11个月内进行了测量。每6周采集一次人类学数据(体长、体重和上臂中部围度(MUAC))以确定营养不良状态,最多采集48周。每12周采集一次尿液样本,用于测量环境性肠道功能障碍(EED)。每12周采集一次粪便样本用于微生物组分析。(B) 来自8名参与者(每个村庄4名)的粪便样本子集。每位参与者(n = 8)在不同时间点提供了5到6个粪便样本。(C) 参与者选择代表了LAZ(年龄-身长Z评分)增长或减少的例子。(D) 优化的高通量分子生物学方法,用于粪便样本处理,包括样本保存、DNA提取和质量控制。(E) 测序文库准备概览,以及使用PB(Pacific Biosciences Revio)、ONT(Oxford nanopore Technologies PromethIOn)和ILMn(Illumina novaSeqX)进行的DNA测序。(F) LR宏基因组研究的元分析,展示了我们的研究如何成为一个独特的资源(蓝色 = ONT,品红色 = PacBio;ONT_vs_PB:0 = 研究仅包含一种数据类型,1 = 研究包含使用ONT和PB生成的样本;纵向:0 = 非纵向,1 = 包含每个样本的多个时间点)。另见图S1、S2、S10和S11。
cMAGs的最佳技术和组装方法
我们通过cMAG组装、概况分析和泛基因组分析等指标对比了测序技术(图S2A--S2D)。我们使用四种组装工具评估了三个平台上的MAG组装性能(LR:metaFlye、metaMDBG;SLR:cloudSPAdes;SR:metaSPAdes)。SR组装未能解决重复区域,表现为前100个contig中的短序列(图2A),这表明SR技术不适合用于恢复完整基因组。Illumina的SLR和SR方法在625 Gbp数据中没有生成任何cMAG,因此后续分析集中在PB和ONT上。PB和ONT的contig比SR长近10倍,特别是在metaMDBG组装方法下,PB的表现优于ONT(图2A;见Mendeley数据中的表S7)
我们在三个参与者样本上(第48周,涵盖两个村庄)进行了深度(>30 Gbp)的LR测序,分别以1、5、10、20、30和40 Gbp的深度进行了子采样,并使用metaFlye和metaMDBG进行了组装。我们使用CheckM2评估了cMAG的数量,设定污染阈值为<5%、contig数=1,完整性阈值为>90%。在每个子采样深度下,PB生成的cMAG数量都超过了ONT,无论使用哪种组装方法(图2B和S3A--S3C;见Mendeley数据中的表S8和S9)。在同一测序技术中比较组装方法时,PB使用metaMDBG组装器生成的cMAG数量更多,而ONT使用metaFlye生成的cMAG数量更多。有趣的是,尽管metaMDBG组装器在处理ONT数据时(与metaFlye相比)表现较好,但我们的结果显示,metaFlye在ONT数据上生成了更多的cMAG,因此我们不建议在ONT数据上使用metaMDBG。
输入数据(Gbp)与生成的cMAG之间的关系在两种方法中是一致的(PB使用metaMDBG和ONT使用metaFlye),我们利用这一关系将cMAG的回收期望建模为测序数据的函数(图2C;见Mendeley数据中的表S9)。平均而言,PB每Gbp生成的cMAG数量比ONT多2.34(标准差±0.79),这一趋势在1--30 Gbp的数据输入中保持一致(图S3D--S3F;见Mendeley数据中的表S9)。使用两种技术成功组装的cMAG跨越了广泛的GC含量范围(图S3G;见Mendeley数据中的表S8)。PB生成的cMAG在完整性上显著高于ONT(Mann-Whitney,p < 0.0001,U = 95832),污染度则显著低于ONT(Mann-Whitney,p < 0.0001,U = 154310),这表明PB的整体质量更高(图S3H和S3I;见Mendeley数据中的表S8)。
低质量的组装通常由于未校正的插入缺失(indels)而包含碎片化的基因。我们使用"Ideal"工具评估基因调用的质量。具有较高错误率的组装,通常来自较低质量的读取,生成的基因较短且碎片化。我们比较了来自相同物种的15个cMAG(来自30 Gbp样本子集A6、D5和H6),发现ONT的metaFlye组装的基因长度比PB的metaMDBG组装要短(图2D;见Mendeley中的表S10)。查询与命中长度的Pearson相关性分析确认,ONT的相关性显著较低(p = 0.02,t = −2.59)(图2E;见Mendeley中的表S10)。尽管SR在我们的研究中没有生成任何cMAG,但我们仍然比较了MAG的质量。在所有样本中,LR组装在所有指标(contig n50、平均长度、contig数量和最大长度)上都优于SR(图S4A--S4D)。即使是ONT和PB的原始读取n50也比SR长约10倍(图S4A)。总体而言,PB与metaMDBG组合生成了最多且质量最高的cMAG,而SR则没有生成任何cMAG。
我们还测试了混合LR数据(PB + ONT > Q20),结合高质量的读取以优化cMAG的回收。在47个样本中的44个中,metaMDBG在cMAG数量上优于metaFlye(一个平局)。对于两种方法,cMAG产量随着输入Gbp的增加而稳定增长,但metaMDBG更高效,产生了46.5%的更多cMAG(986 vs. 673)(图2F;见Mendeley数据中的表S9)。其中,839个是环状的,363个在分类学上是唯一的(GTDB R220),74个是可能的新物种(ANI < 95%)。
我们比较了LR(PB、ONT和混合)和SR(metaSPAdes)技术在生成cMAG时的试剂和消耗品成本,使用了我们的47个样本数据集和Carter等人提供的一个大型ILMn SR数据集(来自351个粪便样本的9,395 Gbp,生成了48,475个MAG和198个cMAG)------这是目前可用的最大SR粪便宏基因组学研究(见Mendeley数据中的表S12)。大多数SR衍生的MAG质量较低或中等。使用输入Gbp与cMAG产量之间的线性关系(图2C;见Mendeley数据中的表S9),我们发现仅使用7.9%的数据(743.1 vs. 9,395 Gbp),我们的方法生成了约5倍的cMAG(986 vs. 198)(图S5A和S5B;见Mendeley数据中的表S13)。仅使用1.4%数据(129.3 Gbp)的PB metaMDBG几乎达到了SR cMAG的输出(197 vs. 208)。LR方法每Gbp生成的cMAG比SR多44--64倍。SR高质量MAG中仅有4.3%是完整的,而LR中的ONT为34%,PB为55%(图S5D;见Mendeley数据中的表S13)。每个cMAG的估算成本为SR 95--2,370(novaSeqX与6000),ONT为25,PB为16(图S5E;见Mendeley数据中的表S13)。虽然多重化可以降低每个样本的成本,但也会降低每个样本的数据量和cMAG产量。当每次运行的样本数超过七个时,ONT才会比PB便宜(图S5F和S5G;见Mendeley数据中的表S14)。
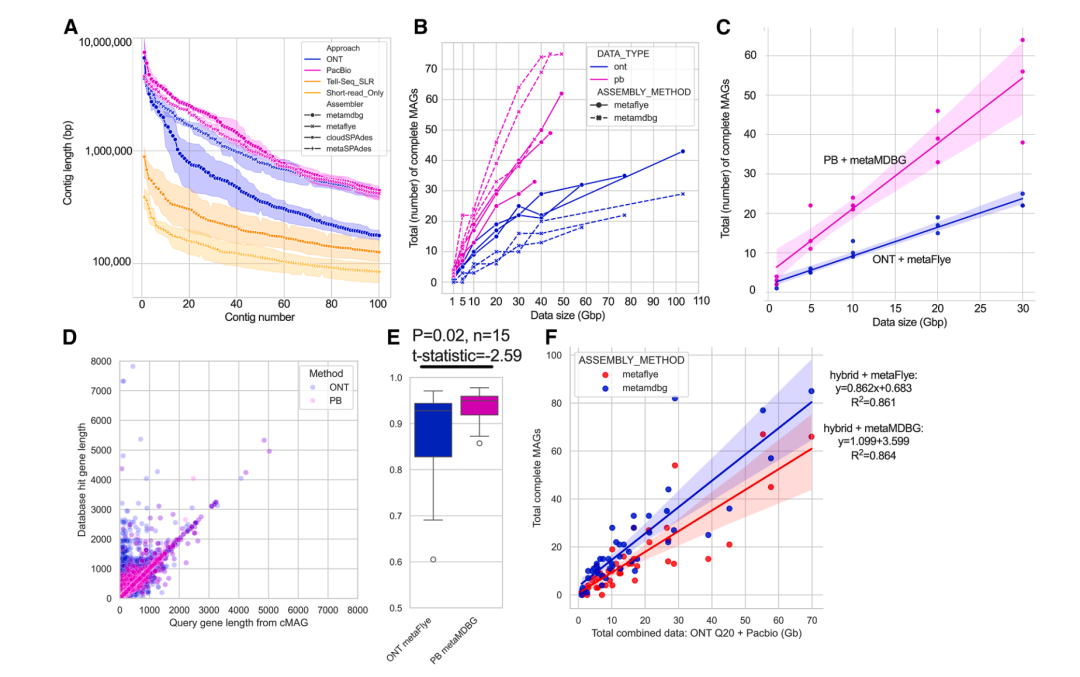
图2 | 不同测序技术和组装方法下的宏基因组组装比较
(A) 不同测序技术(PacBio HiFi "PB" 紫色,Oxford nanopore Technologies Kit14 "ONT" 蓝色,Illumina tell-seq "ILMn" 橙色)和组装方法(metaFlye,metaMDBG,cloudSPAdes,metaSPAdes)下,前100个最长contig的长度比较。
(B) 在儿童年龄最大的第48周时间点,对三位深度测序并进行子样本采样的参与者(A6、D5和H6),比较不同测序技术(PB紫色,ONT蓝色)与组装软件(metaFlye和metaMDBG)在生成完整宏基因组组装基因组(cMAGs)方面的表现。
(C) 建立数据输入(Gbp)与生成的(完整)cMAGs之间的模型,分别为每种技术和组装策略组合的最佳表现(ONT-metaFlye vs. PB-metaMDBG)。
(D) 从三位参与者在30 Gbp深度下使用PB metaMDBG和ONT metaFlye组装获得的15种物种的基因组(cMAG)质量评估。使用Ideal工具比较基因片段长度,查询表示来自给定cMAG的基因长度,而数据库命中表示来自UniProt TREMBL的Diamond索引中的基因长度。
(E) 对30个组装(15种物种:PB和ONT)中的每个进行Pearson相关系数估算。使用配对t检验对每种技术的15个cMAG之间的Pearson相关系数进行配对比较。*p < 0.05。
(F) 混合数据(所有PB HiFi + Q20 ONT)的组装结果(cMAGs)对于所有47个样本。使用线性模型拟合数据,以估算从输入Gbp中恢复cMAGs的数量。
不同技术下粪便微生物群的生态关联一致性
将所有986个cMAG添加到GTDB R220并使用Sourmash进行分析显著提高了分类学的准确性。在用于生成cMAG的重测序样本中,未分类的部分从52.3%(标准差±5.8)下降到16.5%(标准差±5.8),改善幅度为68.5%(图3A;见Mendeley数据中的表S15)。在未用于cMAG构建的新样本中,未分类的读取从53.2%(标准差±7.6)下降到32.4%(标准差±9.8),减少了39%。类似于Kashaf等人通过使用分离基因组改进分类的研究,我们展示了通过添加MAG(在此为cMAG)大大增强了基于k-mer的分类。当将986个cMAG数据库应用于不同LR技术时,PB的未分类读取最少(k = 51,平均值 = 12.3%,标准差±5.8),其次是混合数据(平均值 = 16.1%,标准差±7.2),ILMn(平均值 = 19.2%,标准差±10.0),最后是ONT(平均值 = 40.8%,标准差±3.5)。ONT中较高的未分类部分可能反映了其较低的测序准确度,从而降低了k-mer匹配效率。
接下来,我们评估了测序技术是否会影响分类剖析和多样性指标。对于所有样本,微生物多样性在200--300兆碱基对(Mbp)范围内趋于饱和,因此我们在500 Mbp时进行了稀释。与PB相比,各种方法的丰富度相似(线性模型:ILMn r = 0.8924,ONT r = 0.8766,混合数据 r = 0.9914;p < 0.0001),其中ONT的丰富度略低,ILMn的丰富度略高于PB(图3B;见Mendeley数据中的表S16)。Shannon多样性也一致(ILMn,r = 0.9805;ONT,r = 0.9613;混合数据,r = 0.9920;p < 0.0001)(图3C;见Mendeley数据中的表S16)。ONT的较低准确度可能解释了微小的多样性差异。Beta多样性(Jaccard距离)在不同技术之间没有显示出系统性偏差(图3D),尽管哺乳和生长结果等人口学因素是可以区分的(图3E)。
我们评估了在27个比较中,跨越9个元数据类别的alpha(丰富度、Shannon)和beta多样性(Jaccard),以测试测序技术(ONT、PB、混合数据和ILMn)是否会影响结果。变量是基于先前的证据选择的,预计具有显著性(例如,宿主、村庄和哺乳)或无显著性(例如,性别和饮食组)。为了避免物种组成变化一致性,我们排除了一个仅PB数据表现出显著季节性丰富度差异的实例(p < 0.05,检验统计量 = 4.51)。PB和混合数据检测到了最多的显著关联(14/26),其次是ILMn和ONT(各12个)。与PB或混合数据的一致性为92.3%,适用于ONT和ILMn。不同技术之间的检验统计量是可比的(图3F;见Mendeley数据中的表S17)。所有技术都在丰富度和beta多样性上识别出与线性生长、哺乳和宿主身份的关联。PB独特地检测到了一些关联,例如村庄(ONT未检测到)和采样周(ILMn未检测到)。这些结果支持先前的研究发现,整合多种技术能提高可重复性,并表明LR,特别是PB和混合数据,提供了最高的灵敏度。
鉴于PB(太平洋生物测序)技术的优越性能,我们重新分析了数据集,纳入了三个先前被排除的样本,总计涵盖45个样本和2,194个微生物分类单元(参见 Mendeley中的S18)。在所有时间点中,发现随着线性生长Z评分(LAZ)下降的儿童,粪便微生物丰富度较高(p < 0.01),非哺乳的儿童(p < 0.001),雨季期间(p < 0.05),以及不同参与者之间(p < 0.01),普遍随着年龄的增长(图3G--3K;见Mendeley数据中的表S18)。生长迟滞定义为4个连续的12周间隔中有≥3个间隔ΔLAZ为负(或正)。包括了哺乳状态、季节(11月--4月为雨季)和其他人口学特征(n = 8名参与者,年龄14.4--23.2个月,来自两个村庄)。分析了每个个体的所有时间点。未来的研究应进一步探讨人口学因素如何影响更多个体和不同年龄段的微生物群。
另一方面,粪便微生物生物量在雨季较高(p < 0.05,U = 143,MW)(图3L;见Mendeley数据中的表S18),并且在不同村庄之间存在差异(p < 0.05,U = 364,MW)(图3M;见Mendeley数据中的表S18),但在生长线性、哺乳状态之间没有差异。生物量在参与者之间随时间变化较大,但参与者之间的差异不显著。单一时间点仅捕捉了36%(标准差=22.1)的总alpha多样性,这一数据来自11个月内的5-6个时间点,凸显了纵向采样的价值(图3n;见Mendeley数据中的表S18)。单一时间点仅捕捉了36%(标准差=22.1)的总alpha多样性,来自5-6个时间点,进一步强调了纵向采样的重要性。平均而言,每个参与者在所有时间点出现的分类群占6.5%(标准差=5.5),且没有分类群在所有参与者和时间点之间始终出现,显示出粪便微生物组的高度个体差异性。Beta多样性(Jaccard)与哺乳状态(p = 0.001,F-stat = 7.5,PERMAnOVA9)最强相关,其次是生长迟缓(p = 0.001,F.model = 3.45,Adonis)、线性生长轨迹(p = 0.004,F-stat = 3.09,PERMAnOVA)、个体(p = 0.001,F-stat = 2.7)。村庄(p = 0.038,F-stat = 1.92)和采样周(p = 0.048,F-stat = 1.3)也显著,但关联较弱。总体而言,营养不良表型与多种LR测序方法下的alpha和beta多样性相关。
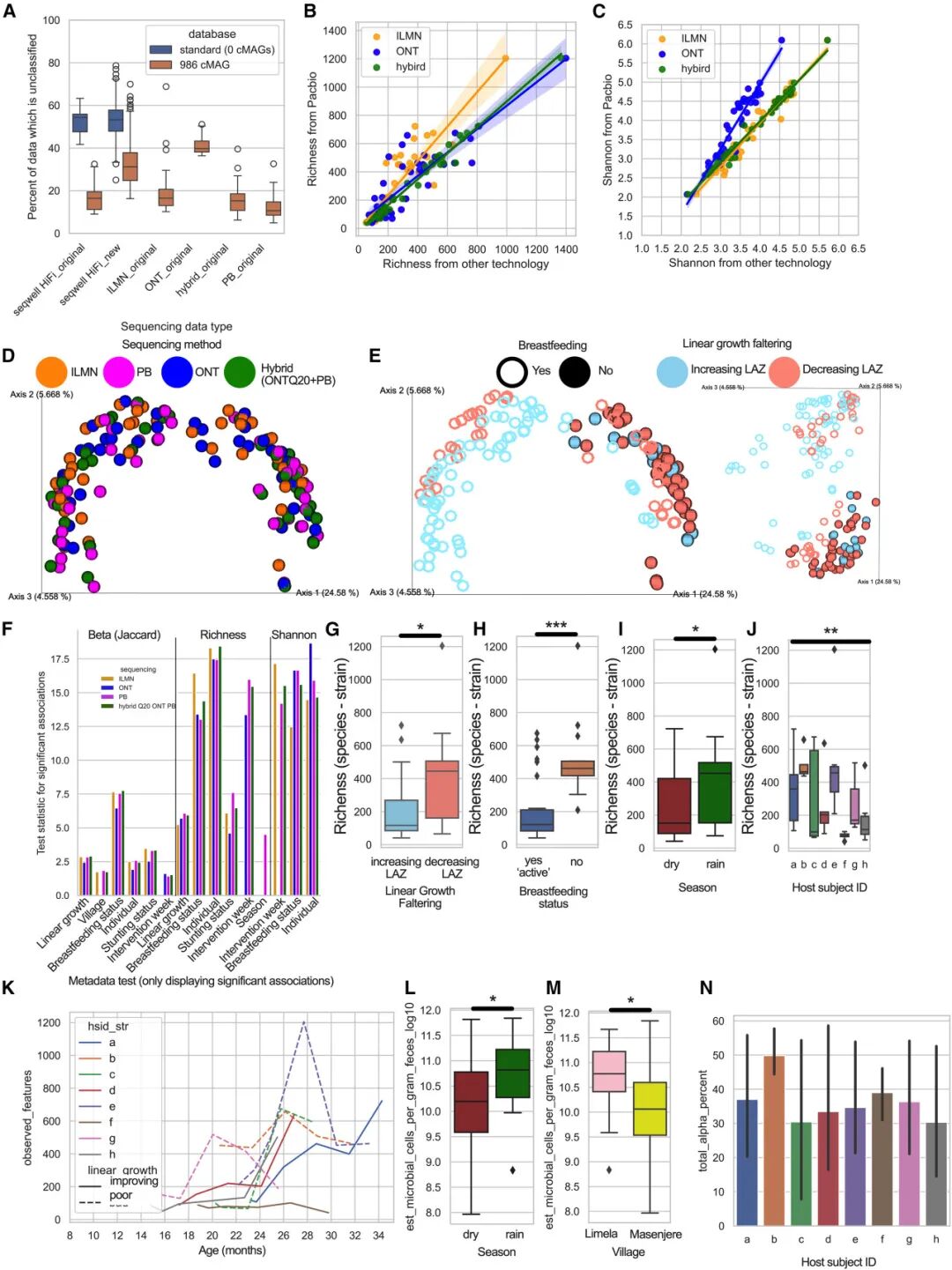
图3 | 通过不同测序技术测量的微生物多样性与生态关联
(A) 将定制修订基因组(GTDB R220 + 986 cMAGs + 47个人类T2T基因组)数据库纳入其中,通过Sourmash改进了宏基因组分类。使用将986个cMAG"微生物"数据库修订为GTDB R220版本,并加入47个人类端粒-端粒(T2T)基因组进行分析,与仅使用GTDB R220标准(0个cMAGs)进行比较。使用Sourmash分类剖析计算的alpha多样性,最低可辨别层次(即菌株或物种)。
(B--E) (B) 丰富度和 (C) Shannon多样性估计值对于42个配对样本,分别在500 Mbp的ILMn、ONT和混合数据下稀释,并与PB进行比较。使用Jaccard距离的PCoA(主坐标分析)图展示了(D)不同测序平台之间的同质性(缺乏偏差)和(E)哺乳状态与线性生长轨迹的强烈影响。
(F) 对18个alpha多样性(丰富度和Shannon的九个元数据类别)和九个beta多样性(Jaccard)比较的统计显著性关联的总结。与PB作为参考(由于PB具有最多显著关联)得出的符合率。仅显示显著(p < 0.05)的关联及相应的检验统计量。
(G--M) 完整的PB HiFi数据(45个样本:B2和C6被排除)在500 Mbp下稀释,展示了与(G)线性生长轨迹、(H)哺乳状态、(I)季节、(J)宿主个体(参与者)和(K)参与者年龄相关的alpha多样性差异,以及(L)季节和(M)村庄之间的微生物生物量差异,并与研究设计参数相关的微生物生物量差异。请注意,alpha多样性是使用Sourmash分类剖析计算的,最低可辨别层次(即菌株或物种)。(N) 单一时间点代表的独特微生物的百分比(一个时间点的丰富度除以所有时间点的独特微生物总数)(平均值,95%置信区间)。
另见图S6。
在扩展的小型验证队列中,识别与生长迟缓相关的特征(物种和菌株)
我们使用机器学习方法识别与线性生长轨迹相关的微生物特征。对39名额外参与者进行了浅层LR宏基因组测序,在五个时间点上使用PacBio HiFi和LongPlex(seqWell)。完整的数据集(39名新参与者 + 8名原始参与者)包括LAZ轨迹、身高矮小状态、性别、年龄、WLZ、哺乳状态、腹泻和12周内的ΔLAZ(见Mendeley数据中的表S19)。合并、稀释和去重后,我们重点分析了在整个过程中身高矮小状态一致的参与者(LAZ < −2 = 矮小;LAZ ≥ −2 = 非矮小),最终得到了41名参与者(22名非矮小,19名矮小)和165个样本。我们评估了Sourmash中的微生物物种/菌株的存在/缺失变异(PAV)(见Mendeley数据中的表S20和S21)。使用随机森林分类器(75:25的训练集/测试集划分)来预测身高矮小状态,确保每个参与者的所有时间点都保留在同一数据集中。此过程重复五次以进行复制。平均分类准确度为62.6%(标准差±10.76,n = 5;范围:51.3%--75.6%),表明尽管样本量较小(22名非矮小与19名矮小),微生物与身高矮小之间存在中等程度的关联(图S6A)。精确度、召回率和F1分数在不同的重复中有所变化(图S6B和S6C),这突显了需要更大样本群体并对年龄、哺乳、地理位置和季节等协变量进行调整。F1分数通常在非矮小参与者中较高,表明对矮小个体的分类较为困难,表现为较高的假阴性。通过五次随机化,18个微生物分类群展示了与身高矮小相关的存在/缺失模式:12个与非矮小参与者相关,6个与矮小参与者相关(图S6D--S6F)。虽然alpha和beta多样性分析表明微生物群落在群体层面与线性生长存在关联,但高个体差异性------在微生物组研究中常见------可能掩盖这些关联。这些发现强调了需要对个体或群体层面进行分层,可能通过低成本的现场生物标志物来指导针对身高矮小、轻度急性营养不良(MAM)和其他形式营养不良的干预。尽管样本量有限,但几种微生物分类群与营养不良表型相关。
微生物基因组稳定性作为线性生长轨迹的预测因子
微生物基因组的多样性可以反映环境的稳定性,但完整的基因组数据很少可得。我们测试了基因组稳定性是否与线性生长轨迹相关,假设稳定的环境可以减少微生物的更替,而压力源则会增加微生物的更替。我们的设计使我们能够评估基因组稳定性与LAZ趋势之间的关系。在8名参与者和986个cMAG中,我们发现了260个基因组,其中73个在个体内重复出现(≥3次)(图4A)。这些基因组中,39个出现在LAZ增加的儿童中,34个出现在LAZ下降的儿童中。LAZ下降的儿童在5.5个月(p = 0.02)、8.5个月(p = 0.0005)和11个月(p = 0.0154,U = 66,Mann-Whitney)时表现出更大的微生物ANI差异------即不稳定性(图4B;见Mendeley数据中的表S22)。
在一个个体的所有时间点上,都存在四个基因组:Collinsella sp900556495(HSID D)、Prevotella copri A(HSID G)、Megasphaera sp900540735(HSID F)和Escherichia coli(HSID D)。只有Dialister sp000434475在所有个体中出现于≥3个时间点,且在LAZ下降的参与者中表现出ANI的一致下降,而在大多数LAZ增加的参与者中则表现为稳定(图4C;见Mendeley数据中的表S22)。这种物种在之前的孟加拉国儿童饮食干预试验中也与LAZ呈正相关。通过纵向完整MAG回收实现的这一分析,突出了研究与生长迟缓相关的基因组动态的新方法,并且可以扩展到其他纵向研究。未来的工作可能会探讨基因的获得/丧失以及驱动基因组差异的分类群。
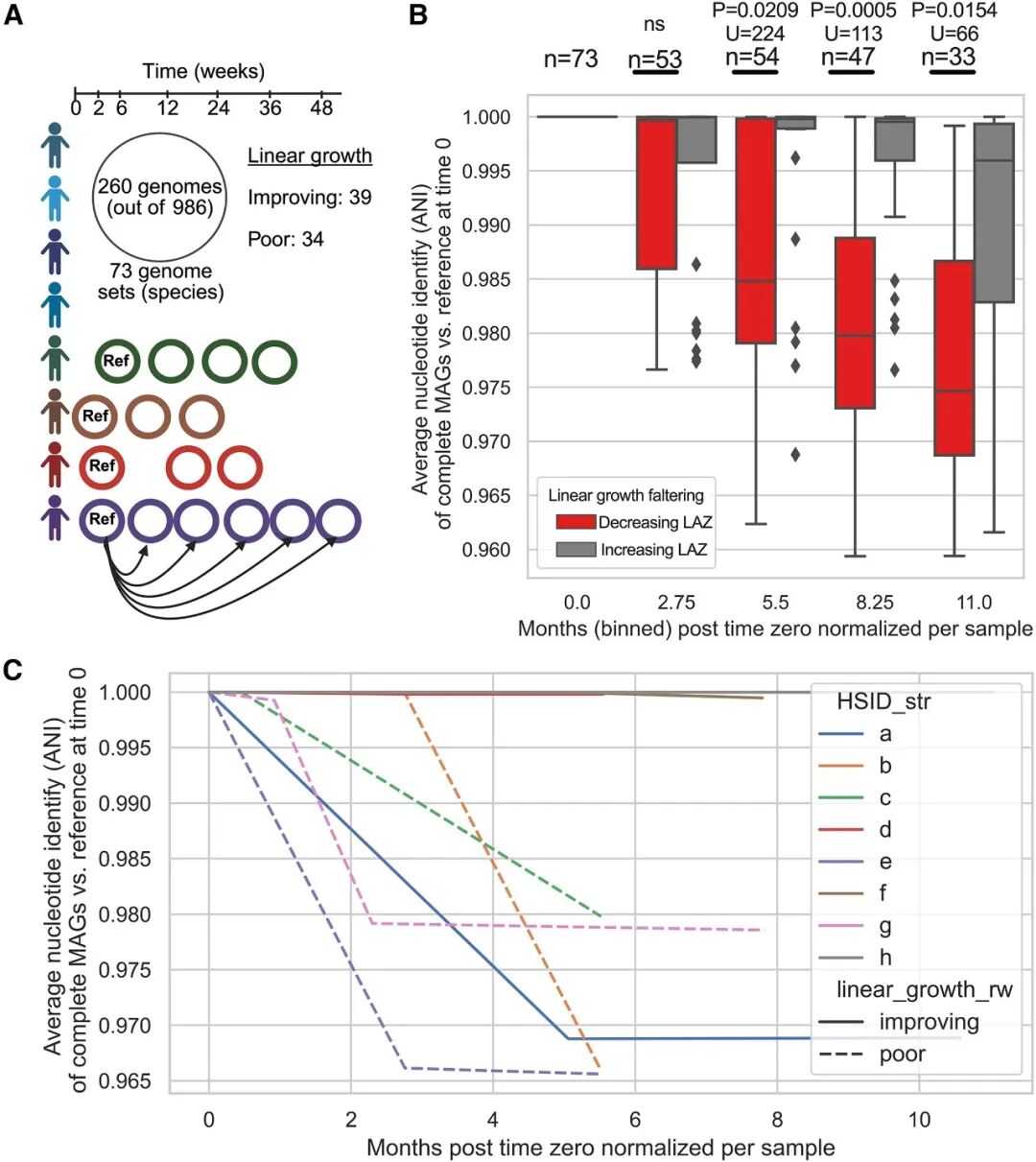
图4 | 时间性微生物基因组不稳定性与线性生长迟缓相关
(A) 从八名参与者的数据集中,共有73个微生物物种在每个个体的至少三个时间点上检测到cMAG代表(AnI > 95%),共获得260个cMAG。基因组相似性(平均核苷酸身份,AnI)是相对于每个基因组组装的第一个时间点计算的。
(B) 比较了在LAZ增加的参与者(n = 39个cMAG集)和LAZ下降的参与者(n = 34个cMAG集)之间,cMAG基因组稳定性随时间的变化。
(C) Dialister sp000434475的基因组分化模式,这是唯一在所有八名参与者中始终被检测到的物种。
cMAG的泛基因组分析揭示了与营养不良和人口学表型的遗传关联
泛基因组分析仅限于我们研究的cMAGs,排除了外部基因组,因为大多数营养不良研究使用的是SR或浅层LR数据。包括不完整或低质量的基因组可能会导致比较结果的偏差。
基于k-mer的泛基因组
微生物组研究通常将分类群或基因丰度与表型关联起来,但可能忽视了常见的"核心"微生物。使用LR数据,我们应用了两种泛基因组分析------全基因组和功能性------以识别与表型相关的菌株级别变异。所有cMAGs都使用了PanKmer,这是一个基于k-mer的工具,用于计算Jaccard距离并评估基因组内容与元数据之间的关联(图5A)。基因变异在普雷沃氏菌属(Prevotella)中最高,其次是Zag111、双歧杆菌(Bifidobacte)、阿加莎杆菌属(Agathobacter)和粪杆菌属(Faecalibacterium),而埃希氏菌属(Escherichia)和巨球形菌属(Megasphaera)的变异最小(图S7A)。我们在来自两个村庄的三名哺乳期参与者的多个时间点中发现了一种可能的新双歧杆菌物种(图S7B)。我们随后分析了丰度最高的10个属(每个属≥20个cMAGs)以检验其与人口统计特征的关联,包括:普雷沃氏菌属(Prevotella)、双歧杆菌属(Bifidobacterium)、粪杆菌属(Faecalibacterium)、阿加莎杆菌属(Agathobacter)、戴阿利斯特杆菌属(Dialister)、Zag111、弯曲杆菌属(Campylobacter)、巨球形菌属(Megasphaera)、拟普雷沃氏菌属(Alloprevotella)以及埃希氏菌属(Escherichia)。(图5B;见Mendeley数据中的表S23)。季节或采样周与任何分类群均未发现关联。然而,普雷沃氏菌属(Prevotella)、双歧杆菌属(Bifidobacterium)、粪杆菌属(Faecalibacterium)、戴阿利斯特杆菌属(Dialister)和拟普雷沃氏菌属(Alloprevotella)的k-mer变异与哺乳状态相关;粪杆菌属(Faecalibacterium)、Zag111和拟普雷沃氏菌属(Alloprevotella)与线性生长相关;而普雷沃氏菌属(Prevotella)、戴阿利斯特杆菌属(Dialister)、巨球形菌属(Megasphaera)和拟普雷沃氏菌属(Alloprevotella)的变异与村庄环境相关(图S7C)。巨球形菌属(Megasphaera)显示出与地理因素最密切的关联,在25个基因组中形成了三个与村庄相对应的独立聚类(其中一个村庄13个,另一村庄12个),这些菌株发现于六名儿童体内(每村各三名),且均接受母乳喂养(图S7D)。
功能性泛基因组
我们对来自前10个丰度最高属的439个cMAGs进行了功能性泛基因组分析,随后进行微生物GWAS,以识别基因型-表型之间的关联。与生长或哺乳的物种数量和关联总结见图5B(见Mendeley数据中的表S23)。使用PIRATE工具识别了基因家族和同源基因。普雷沃氏菌属(Prevotella)具有最高的总功能多样性(26,732个基因家族),而拟普雷沃氏菌属(Alloprevotella)每个基因组的功能多样性最高(29个cMAGs中的14,398个基因家族),其次是埃希氏菌属(Escherichia)(22个cMAGs中的8,009个基因家族)。Zag111的(直系同源)基因簇数量最少(27个cMAGs中的3,824个基因家族)。埃希氏菌属(Escherichia)的旁系同源基因占比最高(18.6%),其次为双歧杆菌属(Bifidobacterium)(13.9%)、普雷沃氏菌属(Prevotella)(13.4%)和粪杆菌属(Faecalibacterium)(13%)。Zag111的同源基因百分比最低(6.7%)(图S8A和S8B;见Mendeley数据中的表S24)。由于人口水平覆盖的不确定性,我们没有评估泛基因组的开放性。基因的存在/缺失矩阵也可能由于组装错误或注释错误而包含错误。同源基因在细菌中的水平基因转移(HGT)中起着重要作用,可能通过基因重复事件或通过分裂与融合产生。分裂或融合事件比基因重复或基因丧失更常见(图S8C;见Mendeley数据中的表S24)。这表明,人类肠道内通过水平基因转移(HGT)实现基因组进化的能力存在差异。在我们研究的类群中,埃希氏菌属(Escherichia)的这种能力是最高的。
接下来,我们对每个十个属进行了微生物GWAS(mGWAS),使用PIRATE的基因存在/缺失输出作为输入,分析了Scoary2(图S9)。我们比较了五个二元元数据类别:线性生长、身高矮小、哺乳状态、季节类型和村庄。十个属中的四个与这些元数据类别存在显著关联(图5C;见Mendeley数据中的表S25)。从这四个属和五个元数据类别中,共识别出了1,912个关联(图5D;见Mendeley数据中的表S26)。大多数关联或命中来自假定蛋白质(1,404/1,912)(见Mendeley数据中的表S26)。
功能性泛基因组分析结果显示,有89个微生物基因或等位基因与良好的线性生长结果显著相关(Scoary2:Fisher检验,FDR 0.05,Benjamini-Hochberg校正),这些基因分布在四个属中:双歧杆菌(1个)、巨球菌(3个)、粪肠球菌(27个)和普雷沃氏菌(58个)(FDR 0.05,Benjamini-Hochberg;见Mendeley数据中的表S26)。其中只有14个基因具有注释的基因名称:arnC、chuR、fim1C、ftsH、fumC、gloB、pknD(2个)、pnbA、rcsC、rhaR、tet(W)、uvrB和xerC。arnC基因是arn操纵子的一部分,使得革兰氏阴性细菌能够修饰脂质A,脂质A是LPS的一部分,从而改变外膜的电荷。LPS通过脂质A的修饰可能帮助细菌通过对阳离子抗微生物肽(CAMPs)、人类防御素和补体蛋白的抗性来逃避宿主免疫,因为它改变了细菌细胞壁的表面电荷。arnC基因在来自良好生长样本的16个巨球菌cMAG中均有发现,但在9个来自差生长样本的巨球菌cMAG中有6个缺失。在普雷沃氏菌cMAG中,arnC基因出现在72个哺乳相关样本中的49个,而在78个非哺乳样本中仅出现在6个。对于P. copri A,30个cMAG中的22个携带arnC基因(见Mendeley数据中的表S26);所有18个普雷沃氏菌sp015074785的cMAG都携带该基因,并且在扩展数据集中,198/201个样本也包含该物种,这表明arnC基因在肠道定殖中具有重要作用,并可能与农村非洲人群(如Hadza族群)相关。
与良好的线性生长结果相关的多个普雷沃氏菌注释基因也与哺乳状态呈正相关,包括chuR、fim1C、fumC、pknD、rcsC和xerC,而gloB则在粪肠球菌中与线性生长和哺乳状态均呈正相关。关于身高矮小的微生物功能性关联,共有169个与巨球菌(1个)、粪肠球菌(3个)和普雷沃氏菌(165个)相关,其中只有35个具有注释的基因名称:arnC(2个)、bshA、btuB、cpdA、cshA、dctP、DNAG、egtB、hcp、lutA、mepA、moeZ、mprA、noc、pnuC、ptk、ptp、rapA(2个)、resA、rfaH、rutC、sasA、sbcC、susC、susF、tagO、tolQ、uppP(2个)、xerC、ydcV、ywqE和zupT。这些细菌基因组中功能性注释的总体低水平使得识别因果关联变得困难,这在该领域是一个重大挑战。尽管我们已经识别出了跨越多个谱系基因组的若干候选基因关联,但在动物模型中进行验证研究仍是至关重要的后续工作。
对四个属分别进行了基于基因存在/缺失(PAV)的主坐标分析(PCoA),结果显示聚类主要遵循GTDB物种分类。我们依据与临床及人口统计学变量的显著关联程度依次分析各属。普雷沃氏菌属(Prevotella)拥有最多的cMAG(150个),涵盖25个物种及若干未分类群(图5E)。普雷沃氏菌的基因组成与母乳喂养关联最强(q = 4.67e⁻¹²,涉及681个基因家族),其次为村庄(q = 1.39e⁻⁵,113个基因家族)、线性生长(q = 0.0024,58个基因家族)、生长迟缓(q = 0.0194,165个基因家族)以及季节(q = 0.0429,1个基因家族)(图5C与5D)。多数物种在LAZ(年龄别身长Z评分)升高与降低的儿童间cMAG数量分布均衡,但Prevotella sp900548535和sp900551275例外,二者各包含5个来自LAZ降低儿童的cMAG,且均未出现在LAZ升高儿童中,这些cMAG普遍与非母乳喂养儿童相关。其他在非母乳喂养儿童中富集的普雷沃氏菌物种包括sp000436035、sp900313215、sp900552675、sp900767615以及普雷沃氏菌粪种(P. stercorea)(图5B)。PERMAnOVA检验(999次置换)发现,普雷沃氏菌属_stercorea (P. stercorea, p = 0.015)与普雷沃氏菌属_copri A(P. copri A, p = 0.022)在基因水平上与线性生长显著关联(图5F、5G);而P. copri A (p = 0.004)和普雷沃氏菌属_hominis(P. hominis, p = 0.033) 则与母乳喂养显著相关(图5H)。
粪肠球菌(Faecalibacterium)在代表性基因组数量排名第三(51个cMAGs),涵盖了12个物种和一个未解析的群体。基因内容与哺乳状态的关联最为显著(q = 0.0056,52个基因家族),其次是线性生长(q = 0.0157,27个基因家族),以及身高矮小(q = 0.0359,3个基因家族)(图5C和5D)。一些物种(如F. prausnitzii E、F、J和F. sp003449675)仅在断奶后出现,而其他物种则仅在哺乳期间出现(图5B)。F. prausnitzii E在LAZ下降的儿童中更为常见(5个 vs. 1个),以及非哺乳儿童中更为常见(6个 vs. 0个)。F. sp900539885在LAZ增加的儿童中略有增多(4个 vs. 1个),所有样本均来自哺乳儿童。F. prausnitzii G是最常见的物种(19个cMAGs),显示出两个与哺乳相关的不同簇,与参与者效应无关(图5I)。它与线性生长(p = 0.01)和哺乳状态(p = 0.001)均显著相关(图5J)。
巨球菌(Megasphaera)在全基因组和功能性泛基因组分析中表现出最强的关联。25个cMAGs包括两个已知物种和一个未解析物种,形成了三个明显的簇。基因内容与村庄的关联最为显著(q = 0.00007,807个基因家族),其次是线性生长(q = 0.0128,3个基因家族)和身高矮小(q = 0.0231,1个基因家族)(图5B--5D)。所有Masenjere cMAGs紧密聚集,而Limela则有两个群体,与PIRATE系统发育树匹配(图5K和S9)。Megasphaera sp900540735在LAZ增加的样本中有更多的cMAG(10个 vs. 3个),但未发现显著的基因差异。所有物种在哺乳参与者中均有所富集(图5B)。
共回收了57个双歧杆菌(Bifidobacterium)cMAGs,使其成为第二大丰富属,包含八个独特物种和一个未解析群体(图5B)。基因内容仅与线性生长相关(q = 0.0167,1个基因家族)(图5C和5D)。双歧杆菌cMAGs形成了四个簇,其中一个簇包含一个推测的新物种(图5L和S7B)。大多数在LAZ增加样本中发现的物种包括B. bifidum(10个 vs. 1个)、B. breve(4个 vs. 1个)、B. infantis(4个 vs. 0个)、B. kashiwanohense_A(9个 vs. 3个)、B. longum(5个 vs. 1个)和未明确物种(8个 vs. 3个)。其他物种的LAZ分布较为均匀或呈下降趋势:B. adolescentis(3个 vs. 2个)、B. pseudocatenulatum(0个 vs. 2个)和B. ruminatium(0个 vs. 1个)。除了B. pseudocatenulatum和B. ruminatium外,所有物种仅在积极哺乳的参与者中发现,这表明这两个物种可能与哺乳无关,并可能与较差的健康结果相关。
另外六个属未与线性生长或哺乳状态表现出整体关联,但在物种级别上存在某些关联。Agathobacter有七个物种加一个未解析物种;A. rectalis在LAZ下降和非哺乳样本中富集(每组6个 vs. 0个),而Agathobacter sp900543445在LAZ增加和非哺乳样本中略有富集(3个 vs. 1个;4个 vs. 0个)。Zag111有三个物种和一个未解析物种,其中Zag111 sp002103105和sp003258735在LAZ下降的样本中略有富集(10个 vs. 3个;4个 vs. 1个)。发现了四个Campylobacter物种,其中C. D sp900539255和C. infans在LAZ下降的样本中略有富集(8个 vs. 4个;7个 vs. 3个)。cdtB基因(CDT毒素的一部分)出现在四个cMAGs中,包括所有C. upsaliensis和一个C. D jejuni。鉴定出六个Alloprevotella物种;sp900541575和sp900542795在LAZ下降的样本中富集(5个 vs. 0个;3个 vs. 0个)以及在非哺乳样本中富集(4个 vs. 1个;3个 vs. 0个)。仅发现了Escherichia属中的E. coli,该物种在哺乳儿童中富集(19个 vs. 3个)(图5B)。
泛基因组分析显示,普雷沃氏菌属(Prevotella)和 粪杆菌属(Faecalibacterium)与线性生长的关联最强,尽管许多物种也与哺乳状态相关,这使得识别主要驱动因素变得复杂。一些物种在增加或下降的LAZ组中有更多的cMAGs。在同时出现在这两种表型中的物种中,Prevotella stercorea与线性生长的关联最强,而Prevotella hominis与哺乳状态的关联最强。P. copri A 和 F. prausnitzii G 则与两者均有关联。先前的研究主要集中在属级丰度变化上,但我们的结果揭示了属内的基因变异与宿主表型的相关性。物种级别的泛基因组分析识别出了与表型强相关的特定物种或基因变种,提供了超越SR宏基因组学的分辨率,推动了微生物组研究的进展。使用SR宏基因组学无法达到这种分辨率,这为微生物组研究的新纪元奠定了基础。
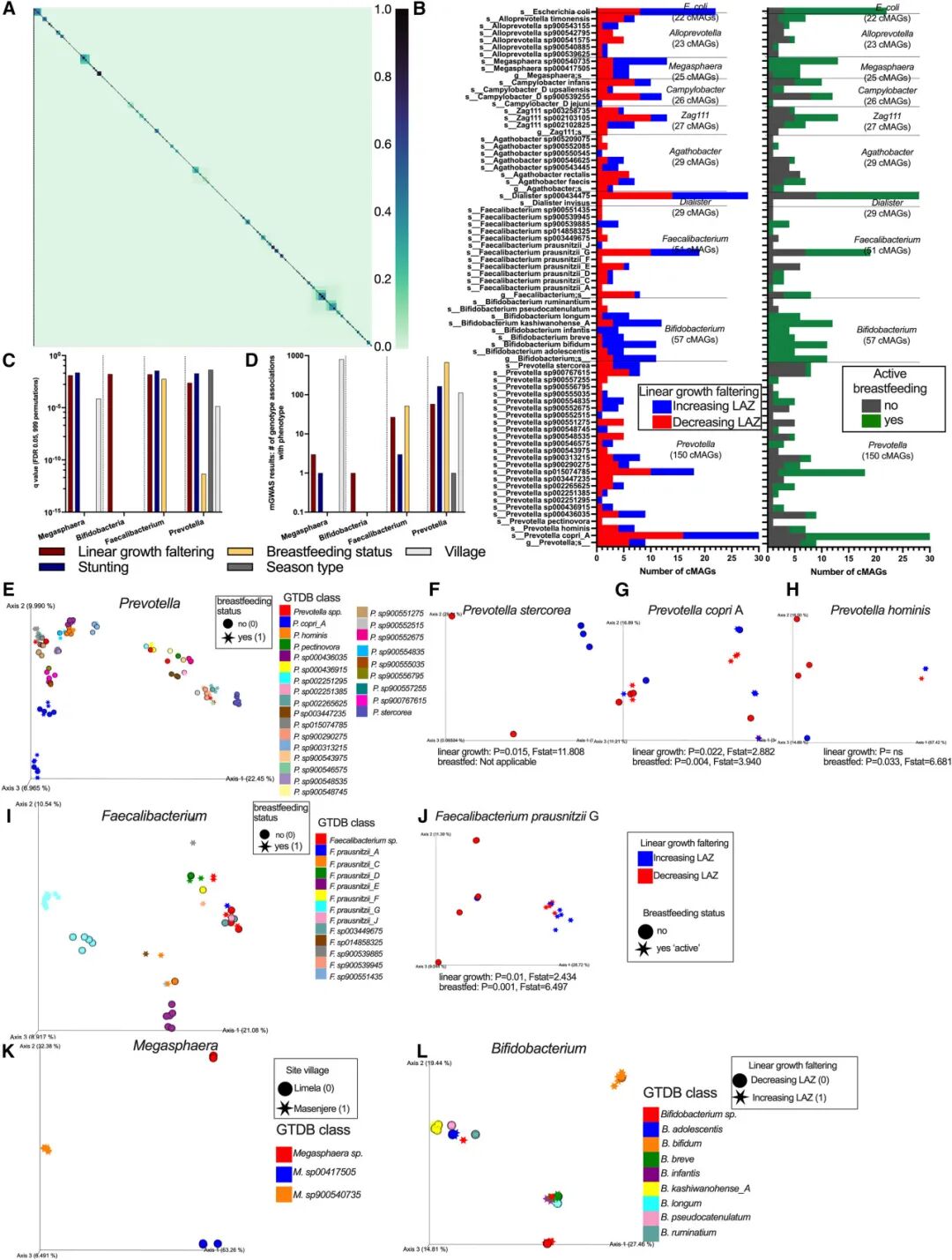
图5 | cMAGs的泛基因组分析揭示了与营养不良和人口学特征相关的属级和物种级基因关联
(A) 使用k-mer泛基因组方法(PanKmer)生成的47个混合LR样本的所有cMAGs的完整泛基因组热图(Jaccard距离)。
(B--D)(B) 列出了从LAZ增加或下降的参与者时间点及哺乳状态中分离出的前十个属的cMAG数量。功能性元泛基因组和微生物GWAS分析使用PIRATE和Scoary2对前十个最丰富的属进行分析:(C) 使用0.05 FDR(Benjamini-Hochberg)筛选的属与表型或实验变量之间的统计显著关联(最佳Fisher q值);
(D) 每个显著关联的微生物基因组广泛关联研究(mGWAS)命中的数量,按四个与显著关联的属进行组织。
(E--L) PCoA图展示了给定属内物种的功能差异。Jaccard距离来源于基因存在/缺失表(PIRATE输出)的cMAGs数据,每个有显著关联的四个属的图形。cMAGs按GTDB分类上色,形状根据每个属不同,但代表与整体最低fq*ep值的关联。
(E) 所有Prevotella cMAGs的PCoA图,按GTDB分类上色,形状根据哺乳状态不同。
(F--H) 在Prevotella给定物种群体中显著关联的PCoA图,包括:(F) P. stercorea(线性生长),(G) P. copri A(线性生长和哺乳状态),(H) P. hominis(哺乳状态)。
(I) 所有Faecalibacterium cMAGs的PCoA图,按GTDB分类上色,形状根据哺乳状态不同。
(J) Faecalibacterium prausnitzii G(显著的线性生长和哺乳状态)的PCoA图。
(K) 所有Megasphaera cMAGs的PCoA图,按GTDB分类上色,形状根据村庄位置不同。
(L) 所有Bifidobacterium cMAGs的PCoA图,按GTDB分类上色,形状不同。
另见图S7、S8和S9。
噬菌体基因组整合与营养不良的关联
HGT(水平基因转移)是细菌基因组进化的关键驱动因素,但在宏基因组中的SR组装限制了其研究。通过使用来自纵向样本的环状MAGs,我们追踪了HGT和结构变异(SVs)在个体菌株中的时间变化,重点关注前噬菌体------这些细菌病毒会整合到宿主基因组中。我们从86个菌株中识别出204个环状MAGs,这些菌株在多个时间点上显示出前噬菌体整合。尽管大多数前噬菌体在11个月的研究中保持稳定,但许多前噬菌体出现后又消失(图6A;详细信息见Mendeley数据中的表S27)。染色体水平的视图(例如Megasphaera、Prevotella、Faecalibacterium、Duodenibacillus和Alloprevotella)揭示了与前噬菌体活动相关的大型结构变异(图6B;详细信息见Mendeley数据中的表S27)。
我们评估了在不同二元表型(线性生长、母乳喂养、村庄、季节和性别)下每个环状细菌基因组的噬菌体计数。平均而言,每个基因组有2.93个噬菌体整合(标准差=1.96;范围:0-14)。母乳喂养的儿童噬菌体整合显著更多(p = 0.001,U = 67983),女性的噬菌体计数高于男性(p = 0.012,U = 52653)(图6C--6E;见Mendeley数据中的表S27)。随着LAZ时间点的增加,噬菌体计数趋势较高,但未达到显著性(p = 0.17)。季节或村庄与噬菌体整合没有发现显著关联。噬菌体整合水平在时间和参与者之间保持稳定(图6F和6G),但在细菌目之间有所不同(图6H)。在十大属中,Escherichia(6.93)、Alloprevotella(5.1)和Faecalibacterium(4.16)具有最高的噬菌体计数;而弯曲菌的计数最低(每个基因组1个)(图6I)。在Faecalibacterium中,随着LAZ增加,噬菌体整合较高(p = 0.018,U = 311.5),并且不同物种之间存在差异(图6J和6K)。在肠腔中富集的温和噬菌体,可能对婴儿的屏障功能和免疫防御起到支持作用。
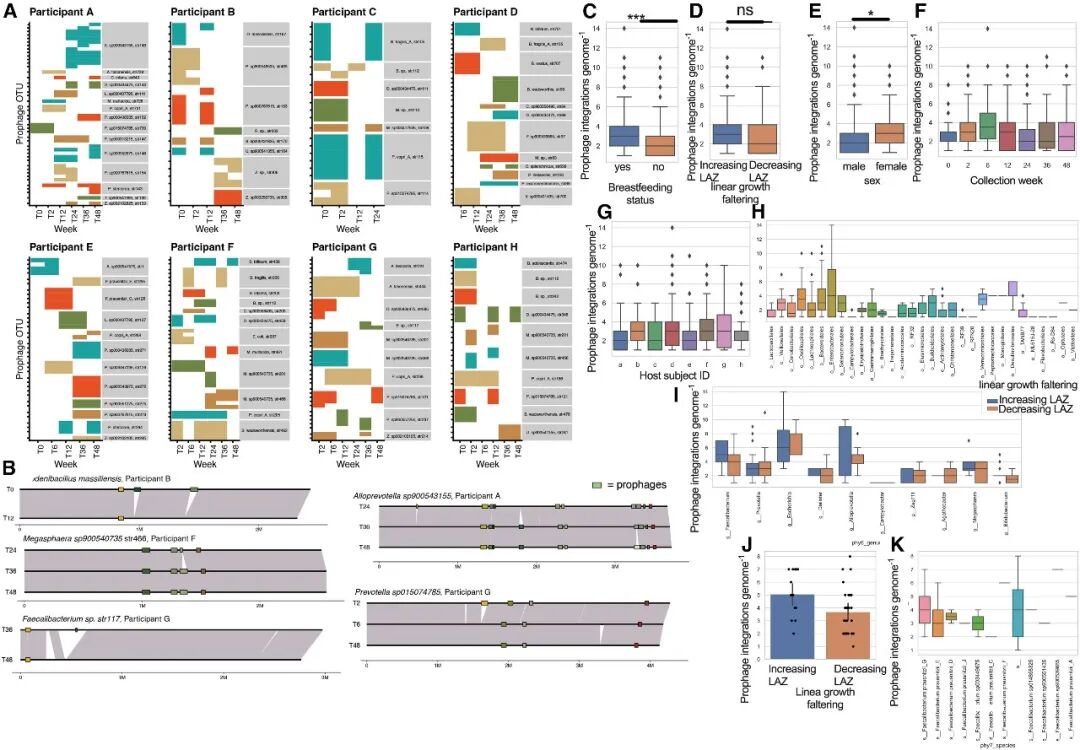
图6 | 环状微生物基因组的溶源噬菌体分析
(A) 在每个参与者中出现多次且具有一个或多个前噬菌体的细菌菌株,被绘制为存在-缺失变异(PAV)矩阵,以记录基因组中的前噬菌体。大多数前噬菌体在时间点之间保持稳定。然而,也可以观察到前噬菌体的丧失、获得或交换的情况。
(B--E) (B)五组纵向环状MAGs的基因组对齐图,用于记录时间点之间的结构变异(SVs)。前噬菌体的坐标标记为彩色框。在这些情况下,大多数结构变异可以归因于前噬菌体的获得或丧失。针对不同表型的环状细菌MAGs中每个基因组的噬菌体整合事件的比较,包括(A)母乳喂养状态,(B)线性生长,(C)性别,(D)采样周次,和(E)参与者。
(F 和 G) (F)在所有分类群中的每个基因组中噬菌体整合数的变异,(G)在十大属中的变异。
(H) Faecalibacterium基因组与线性生长的关联及Faecalibacterium物种之间的变异。
讨论Disscussion
儿童营养不良仍然是全球重大健康挑战,导致45%的五岁以下儿童死亡。通过优化的长读长(LR)测序流程,我们从马拉维的47个粪便样本中生成了986个完整的微生物基因组(cMAGs),这些样本是在11个月内采集的。尽管我们只使用了7.9%的数据,我们的数据集生成的cMAGs数量是一项短读长(SR)研究的5倍,并且提高了68%的分类精度。
我们对cMAGs进行了全基因组和功能泛基因组分析,以识别与营养不良相关表型(如线性生长停滞、发育迟缓和母乳喂养状态)相关的属。这些关联可能源于不同菌株和物种在对立的表型组中占主导地位(即丰度差异),正如在不同村庄间观察到的巨球形菌属(Megasphaera)一样,另一种可能性是,物种或菌株在不同群体之间共享,但基因内容或等位基因变异不同------这些模式提示了选择作用。这后一种情况更为常见,并且只有通过我们采用的非靶向宏基因组方法才能检测到。
微生物全基因组关联(mGWAS)分析识别了物种/菌株与营养不良相关表型之间的功能联系。泛基因组结果显示,尽管样本量较小,但在双歧杆菌属(Bifidobacterium)、巨球形菌属(Megasphaera)、粪杆菌属(Faecalibacterium)和普雷沃氏菌属(Prevotella)中发现了与线性生长相关的遗传关联。发育迟缓也与巨球形菌属(Megasphaera)、粪杆菌属(Faecalibacterium)和普雷沃氏菌属(Prevotella)中的基因水平变异相关。弯曲杆菌(Campylobacter)基因组在LAZ下降的儿童中更为常见。在18个关键的生长预测分类群中,七个是普雷沃氏菌属(Prevotella)菌株,其中既包括正向关联也包括负向关联,这突显了物种或菌株水平分辨率的必要性。巨球形菌属(Megasphaera)物种的基因内容与地理位置(村庄)密切相关。巨球形菌属(Megasphaera)可引起细菌性阴道病,与HIV风险相关,并且通常在女性中更为丰富。我们研究中,巨球形菌属(Megasphaera)在75%的女性和50%的男性中出现。
物种水平的泛基因组分析显示,普雷沃氏菌属(Prevotella)与母乳喂养的关联最强,其次是与线性生长的关联(图5C和5D)。几种普雷沃氏菌属(Prevotella)物种在增加或减少的LAZ组中富集,并在物种内表现出基因组差异。例如,P. copri A基因组在母乳喂养和生长方面有所不同(图5G),P. stercorea与生长相关,P. hominis与母乳喂养相关(图5F和5H)。在粪杆菌属(Faecalibacterium)中,F. prausnitzii G基因组与线性生长和母乳喂养均相关,并在来自LAZ增加的儿童的微生物基因组中表现出更高的噬菌体整合。Faecalibacterium还与克罗恩病和炎症性肠病(IBD)等胃肠疾病相关。Faecalibacterium在肠道疾病患者中通常较为缺乏,是一个关键的丁酸盐生产者,支持肠道屏障功能。基因组中更高的噬菌体多样性可能增强抗病毒防御。未来的研究应探讨,是否具有更多噬菌体整合的微生物是长期肠道居民,并且对微生物继代更具抵抗力。我们的泛基因组分析突显了来自混合群体的完整基因组如何揭示与表型相关的亚物种级别的遗传适应。
我们的纵向基因组分析显示,LAZ增加的儿童具有稳定的微生物基因组,而LAZ下降的儿童则表现出更大的基因组变化,尤其是在5个月后------这表明基因组稳定性可能成为评估肠道健康的新方法。纵向研究设计与重复采样事件对应健康轨迹,应优先于横断面设计。单一时间点仅捕捉到在11个月期间5-6个样本所见的总α多样性的约36%。LAZ下降的儿童具有更高的微生物丰富度,并受季节(雨季较高)、年龄(较大的婴儿较高)和母乳喂养状态(断奶后较高)影响。这些趋势需要在更大规模的队列中验证,特别是季节性效应。我们的研究是首次将微生物群动态与生长轨迹联系起来的重要研究之一------这是一个重要的区别,因为儿童可能是发育迟缓但正在改善(LAZ增加),或非发育迟缓但处于风险中(LAZ下降)。Robertson等人发现,在赞比亚的一个更年轻队列(1-18个月龄)中,微生物功能特征(以及程度稍轻的分类学特征)对线性生长具有中等程度的预测能力。个体间微生物群落的巨大差异表明,未来需要进一步研究婴儿微生物群的来源(如父亲的影响)及其环境与生物学驱动因素。
除了为研究儿童营养不良提供关键资源外,我们资源中的免费开放数据集对于那些寻求开发LR分析工具的研究者也具有重要价值。近年来,LR测序取得了显著进展。由于开发了14号文库制备套件、R10.4流动池和孔径率,ONT平台的碱基呼叫准确性得到了提高。随着"Revio"仪器的发布,PB的整体产量和通量也有所增加,该仪器每个SMRT单元的ZMW数量从800万个增加到2500万个,并且能够运行更多的SMRT单元(从1个增加到8个),相较于前代仪器Sequel IIe。由于这些进展,许多新的软件工具已被开发并将继续开发,用于LR宏基因组学应用,包括组装、分类剖析和甲基化检测等。
单分子测序(PB或ONT)在临床应用中的一个关键挑战是跨平台的可重复性:一个平台的发现能否在另一个平台上得到复制?我们的数据集是迄今为止最全面的LR宏基因组学数据集(图1F)。此前的比较研究通常使用过时的技术,采用虚拟群落而非"真实样本",并且样本量通常较小(少于三个)。例如,Bhatt等人使用了仅两个配对样本和较旧的平台(ONT R9.4,PB Sequel),而另一项使用ONT R10.4的研究仅测试了一个样本(厌氧消化器),并使用了过时的PB化学试剂(SMRTbell 1.0)。另一项较新的研究比较了两个粪便样本,产量较低(PB:14.7 Gbp,ONT:17.5 Gbp),限制了性能分析。在我们的分析中,92%的显著变量在LR和SR技术之间是一致的,PB检测到更多的关联,表明其敏感性更高。
LR和SR技术都识别出了与婴儿营养不良相关的微生物关联,突显了将粪便宏基因组学应用于临床全球健康的机会。ONT可能使得在现场进行菌株解析的宏基因组测序成为可能,结合机器学习,能够支持快速的传染病诊断和营养不良等疾病的风险预测。我们使用我们的数据集比较了不同平台之间的测序性能、组装、成本和剖析。ONT PromethIOn的平均产量高于PB(78.11 vs. 42.24 Gbp),但可用数据较少(71.5% vs. 93.2%),导致净输出相似。尽管ONT宣传每个流动池可获得高达290 Gbp的数据(相比PB的90 Gbp),但PB具有更高的平均、媒体和n50读长。ONT的测序长度通常远低于TapeStation和Femtopulse的DNA长度测量值。一个可能的解释是,较短的片段更倾向于在ONT平台上进行测序。PB的读长质量比ONT高近100倍(更准确),这有助于更好的cMAG质量。较高的读长质量提高了组装和注释的准确性,而这些对于准确的泛基因组比较至关重要。使用metaMDBG,PB生成的完整、低污染的cMAG比使用metaFlye的ONT多2.3倍。每个cMAG的成本对于PB(16美元)和ONT(25美元)低于ILMn(95美元至237美元),因为PB和ONT的效率更高(每Gbp生成44至64倍更多的cMAG)。尽管LR平台的每个样本成本较高,但通过多重化可以将其降低至50至150美元,新版PB工作流程接近每样本40美元(针对96个样本)。相比之下,高通量SR宏基因组学的每样本成本可以低至10至15美元。以往的成本比较仅关注过时的每Gbp费用;我们的分析侧重于cMAG的生成。ONT也可能对微生物定量有用,因为我们已将我们的生物量估算方法适配到了ONT,使用了RBK文库制备。
LR宏基因组学终于解锁了从混合样本中大规模恢复完整微生物基因组的能力。我们展示了这一方法如今在大量样本中是可行的,从而使得基于宏基因组样本的宏-泛基因组和mGWAS分析成为可能。结合纵向研究设计,我们展示了微生物进化和基因变化如何与儿童营养不良相关的不同健康轨迹(特别是发育迟缓)相关联。这种新方法将使微生物生态学和进化领域能够提出全新的研究问题,并推动解决全球健康挑战的新发现。我们的研究展示了将LR宏基因组学应用于解决影响全球超过1.5亿儿童的儿童营养不良这一紧迫问题的可行性。值得注意的是,这些相同的方法在大流行监测、传染病诊断(如结核病)和抗生素耐药性监测等方面具有广泛的转化应用潜力。我们的研究结果强调,跨学科的微生物组研究将从cMAGs的生成中获得深远的利益,能够实现高分辨率的比较基因组学、泛基因组分析以及新一代精准分析得以实现。
作者简介
Jeremiah J. Minich(第一/通讯作者)

美国拉荷亚索尔克生物研究所nOMIS--Salk研究员。致力于理解鱼类消费如何帮助减轻或预防低收入和中等收入国家营养不良的负担。作为nOMIS研究员,他将研究肠道微生物组在婴儿营养不良中的作用。营养不良影响全球超过1.49亿儿童,是饮食不足、微量营养素缺乏、肠道功能障碍、肠道炎症和微生物组紊乱共同导致的。Minich正在开发新型高通量长读长测序方法,用于测量和比较微生物组。通过将这些先进的测序方法与机器学习结合,杰克旨在从非洲马拉维的回顾性队列中发现预测婴儿疾病进展的生物标志物。
Rob Knight(通讯作者)

微生物组创新中心的创始主任,同时也是加州大学圣地亚哥分校儿科、生物工程和计算机科学与工程的教授。他是沃尔夫家族微生物组研究捐赠讲席教授,同时也是美国科学促进会和美国微生物学会的会士。他因其微生物组研究荣获2019年美国国立卫生研究院主任先锋奖,并于2017年获得马斯里奖,该奖常被视为诺贝尔奖的预测者。他是《跟随肠道:微小微生物的巨大影响》(西蒙与舒斯特,2015年)的作者,合著了《土壤是好事:细菌对你孩子免疫系统发展的优势》(圣马丁出版社,2017年),并发表了700多篇科学文章。他于2014年发表了一场TED演讲,观看次数超过210万次。他的实验室开发了许多实现高通量微生物组科学的软件工具和实验室技术,包括QIIME(微生物生态定量洞察)流程(截至本文被引用超过3万次)和UniFrac(被引用超过1万次)。他是地球微生物组项目、美国肠道项目以及利用地球地下微生物DnA指导油田决策的Biota公司联合创始人。他的研究将微生物与多种健康状况联系起来,包括肥胖和炎症性肠病;加深了我们对从海洋到苔原等环境中微生物的理解;并使高通量测序技术能够惠及全球数千名研究人员。
Mark J. Manary (通讯作者)

马克·马纳里医学博士是华盛顿大学儿科海伦·B·罗伯森教授,花生酱项目创始人,并开发了一种革命性的花生即用治疗食品(RUTF)。RUTF已成功应用于全球营养不良儿童治疗。他目前与花生创新实验室合作的研究,将RUTF应用于马拉维治疗中度不良营养因子孕妇,以防止儿童发育迟缓。他每年有11个月居住和工作在马拉维,并在撒哈拉以南非洲从事工作超过20年。
Todd P. Michael(通讯作者)

Todd P. Michael是基因组测序和分析领域的顶尖研究者,目前担任索尔克生物研究所的研究教授及加州大学圣地亚哥分校兼职教授。迈克尔博士拥有涵盖多个重要职位的杰出职业生涯,包括J. Craig Venter研究所(JCVI)的信息学教授兼主任,以及孟山都基因组中心主任,确立了自己在植物基因组学和生物信息学领域的先驱地位。迈克尔博士在达特茅斯学院获得分子与细胞生物学博士学位,师从C. Robertson McClung博士,此前他在弗吉尼亚大学获得生物学学士学位,师从迈克尔·蒂姆科。他在索尔克研究所与著名植物生物学家乔安妮·乔里的博士后合作为他丰富的科研生涯奠定了基础,在此期间他发表了100多篇论文,累计被引用超过21,000次。迈克尔博士的研究为理解植物基因组结构做出了重要贡献,特别是通过研究独特且专门化的植物,这些植物为适应策略提供了见解。他在全基因组工具和长读长测序技术方面的创新工作,推动了对植物基因组组织及其适应特定环境条件的理解。迈克尔博士的工作持续推动我们对植物生物学理解的边界,对农业、保护和气候韧性产生深远影响。
翻译:韩维,中国农科院油料所博士在读
审核:朱志豪,广东医科大学,基因组所联合博士后
终审:刘永鑫,中国农科院基因组所,研究员/博导
排版:杨海飞,青岛农业大学,基因组所联培硕士在读
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
iMeta高引 fastp PhyloSuite ImageGP2iNAP2 ggClusterNet2
iMeta工具 SangerBox2 美吉2024 OmicStudioWekemo OmicShare
iMeta综述 高脂饮食菌群 发酵中药 口腔菌群 微塑料 癌症 宿主代谢
10000+:扩增子EasyAmplicon 比较基因组JCVI 序列分析SeqKit2 维恩图EVenn
iMetaOmics高引 猪微生物组 16S扩增子综述 易扩增子(EasyAmplicon)
点击阅读原文