系列文章目录
文章目录
- 系列文章目录
- 📌1.材料专家------用于材料发现的人工智能
- [📌 2 适用于元素周期表的通用图深度学习原子间势](#📌 2 适用于元素周期表的通用图深度学习原子间势)
- 📌3.利用少样本机器学习实现电子显微镜数据的快速灵活分割
📌1.材料专家------用于材料发现的人工智能
Materials Expert-Artificial Intelligence for materials discovery communications materials
主要贡献
提出了一种新的机器学习框架 ME-AI,该框架通过结合人类专家的直觉和实验数据,发掘新的描述符来预测材料的性质。以下是这篇文章的主要贡献和创新点:
核心贡献:
-
ME-AI框架: 这个机器学习模型将专家策划的数据与实验数据结合,通过从中提取定量描述符来预测材料性质,特别是针对 拓扑半金属(TSMs) 在方形网状材料中的识别。
-
专家知识与机器学习的结合: 文章的创新之处在于,将材料专家的直觉和经验通过实验数据"封装"成机器学习模型的输入,从而揭示出能有效预测材料性质的新的量化描述符。
主要创新点:
-
自动发现新的描述符: 在方形网状材料的数据库上,ME-AI不仅重新发现了传统的 t因子(t-factor),还发现了四个新的描述符。其中一个新的描述符与化学中的超价化学(hypervalency)概念相关,能够帮助识别拓扑半金属。
-
拓扑绝缘体预测: 通过将ME-AI框架应用于由专家标记的879个方形网状材料,模型能够有效地识别拓扑半金属,并且能够将这种识别能力扩展到其他材料结构,如岩盐结构,展示了模型的普适性和迁移能力。
-
新的化学理解: 通过机器学习提取的描述符,文章揭示了 超价化学 对于拓扑半金属形成的关键作用,特别是 χ_sq*fcc 描述符,它反映了在方形网状材料中原子间的共价键合,能够帮助理解这些材料的电子结构。
-
高准确率和泛化能力: 该模型通过高斯过程回归(Gaussian Process Regression,GPR)进行了训练,展示了极高的准确性(超过98%),并且能够从方形网状材料推广到其他结构类型(如岩盐结构),进一步证明了其通用性和稳定性。
摘要
材料数据库的发展为挖掘可预测新兴性质的描述符创造了机遇,但多数研究依赖高通量从头算计算,这类计算结果可能与实验存在偏差。实验研究者则依赖实践积累的直觉。我们提出"材料专家-人工智能"(ME-AI)------这是一个机器学习框架,可将此类直觉转化为从机器测量数据中提取的定量可解释描述符。
我们以12个实验特征描述的879种方网化合物为数据集,训练了带有化学感知核的狄利克雷基高斯过程模型。ME-AI不仅复现了识别拓扑半金属(TSM)的已知专家规则,还揭示了"超价性"是这类体系的关键化学调控因素;更值得注意的是,仅用方网TSM数据训练的模型,能正确分类岩盐结构中的拓扑绝缘体,体现出良好的迁移性。
该框架可与电子结构理论互补,能随数据库扩容而扩展,嵌入了专家知识、提供了可解释判据,还能指导靶向合成,从而加速不同化学家族的材料发现与快速实验验证。
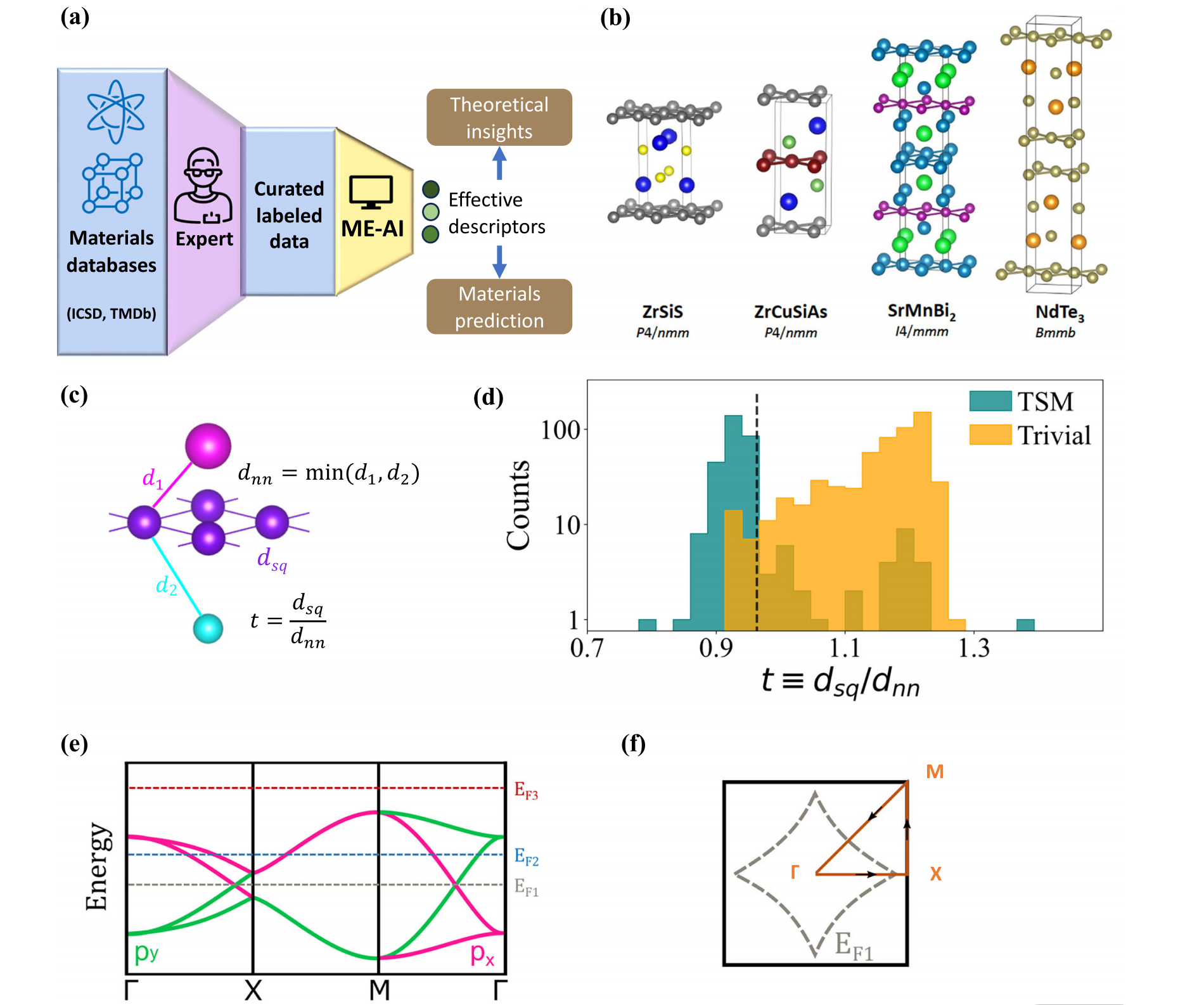
图1 | 用于方网材料中拓扑半金属研究的"材料专家-人工智能"(ME-AI)
a a a ME-AI的概念框架。
b b b 部分二维方网材料的晶体结构。
c c c 三维方网结构:面外的品红色与青色原子到方网平面(紫色)的距离分别为 d 1 d_1 d1和 d 2 d_2 d2;容忍因子定义为 t ≡ d sq / d nn t \equiv d_{\text{sq}}/d_{\text{nn}} t≡dsq/dnn,其中 d nn ≡ min ( d 1 , d 2 ) d_{\text{nn}} \equiv \min(d_1, d_2) dnn≡min(d1,d2)代表面外最近邻间距。
d 数据集中标记为拓扑半金属(青绿色)与平庸材料(橙色)的化合物的容忍因子 t t t分布(其中 t ≡ d sq / d nn t \equiv d_{\text{sq}}/d_{\text{nn}} t≡dsq/dnn, d nn d_{\text{nn}} dnn为面外最近邻间距);我们发现 t ≈ 0.96 t \approx 0.96 t≈0.96(虚线)能以最高准确率区分拓扑半金属与平庸材料。
e e e 方网形成过程中假设的 p x p_x px(粉色)与 p y p_y py(绿色)轨道的紧束缚能带结构,沿f图所示路径绘制;图中展示了两处伴随能带反转的对称保护交叉点;费米能级的位置决定了材料是能带绝缘体( E F 3 E_{\text{F}3} EF3)、金属( E F 2 E_{\text{F}2} EF2)还是拓扑半金属( E F 1 E_{\text{F}1} EF1)。
f f f 布里渊区( B Z BZ BZ):当费米能级调至穿过节线时,节线即为费米面。
正文
过去十年的全球努力构建了庞大的材料数据库¹⁻¹¹,让研究者可查询单个条目的关联数据。这个"材料数据时代"为通过推导条目间的关联与趋势来发现物理性质描述符,提供了前所未有的机遇。历史上最成功的案例是门捷列夫:他通过元素周期表的经验趋势预测了元素材料的性质。但发现非元素材料的拓扑特征仍极具挑战------这类材料的组合相空间与特征空间的高维度,已超出人工探索的范围。
机器学习(ML)工具的普及推动了近期进展²³⁻²⁴:研究者通过扩大从头算计算与合成规模筛选稳定材料。但从头算预测的局限在于,无法囊括实验研究者多年积累的直觉与经验。我们的目标是将专家(ME)的直觉(辅以材料性质标注)转化为ML模型,让ME-AI从实验数据中提取信息、明确这些直觉(见图1a)。
拓扑半金属(TSM)因新奇物理性质与应用潜力受关注²⁵⁻²⁷,其核心特征是"自旋简并能带交叉的节线"与"费米能级调控至节线处"。传统TSM识别需分析材料对称性、推导能带表示,但我们观察到:化学直觉可助力在二维"方网材料"中预测TSM(这类材料是含2D中心方网的固体,见图1b、c)。基于这一直觉,我们在文献26、27中引入"容忍因子(t因子)"------定义为"方网晶格间距与面外最近邻间距的比值"(图1c),它能有效区分TSM(t值较小)与平庸材料(图1d)。
为保证完整性,我们先回顾t因子的结构逻辑:方网材料的二维方网基序层间堆叠着其他原子;若通过二维紧束缚模型(将面外原子投影至方网中心、方网原子pₓ/pᵧ轨道部分填充)最小化面外原子的影响,其能带结构会预测出"受对称性保护的拓扑节线"(图1e、f)。若该模型成立且费米能级穿过节线,这类材料就是TSM²⁹。这启发我们用t因子(dₛq/dₙₙ)量化与二维方网平面的偏离程度,但3D晶体的低能电子性质还取决于电子填充、干扰能带与费米能级位置³⁰。
本研究核心
我们开发的ME-AI框架,利用实验整理的专家直觉挖掘TSM的定量描述符:将其应用于879种方网化合物(12个核心特征)后,ME-AI不仅识别出"容忍因子",还发现了4个新描述符(其中一个纯原子描述符契合"超价性、津特耳线"等经典化学概念)。它不仅预测能力优异,还能迁移至岩盐结构中识别拓扑绝缘体------这凸显了"人类专家知识+人工智能"在靶向材料发现中的潜力。
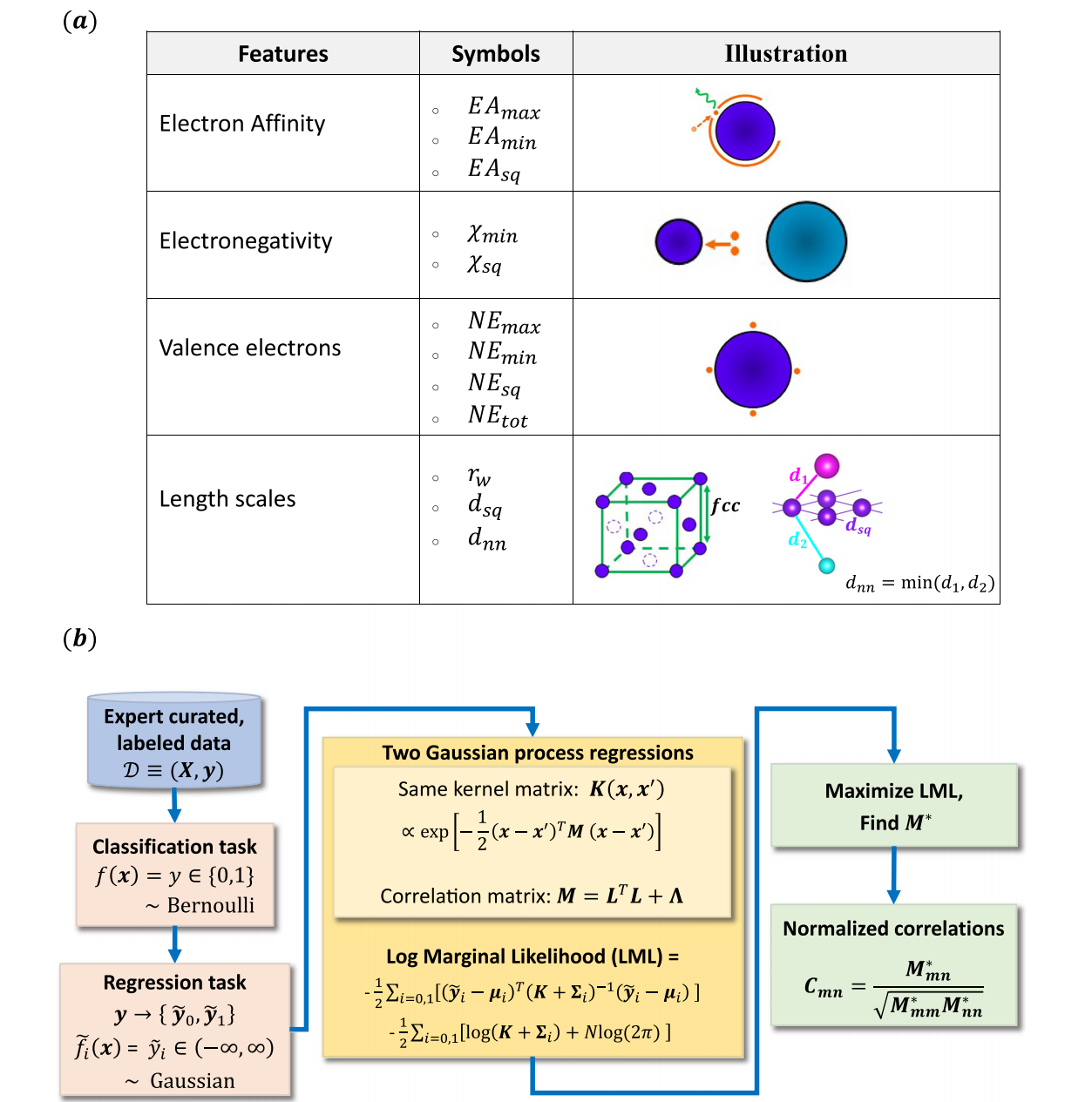
图2 | ME-AI的机器学习方案
a 核心特征。下标 max \text{max} max、 min \text{min} min、 sq \text{sq} sq分别代表最大值、最小值、方网元素对应的值;下标 tot \text{tot} tot代表基于化学计量信息的总电子数。我们省略了 χ max \chi_{\text{max}} χmax,因其与 χ sq \chi_{\text{sq}} χsq的协方差较高。
b ME-AI的流程图解。我们首先构建预处理并标注的数据集 D ≡ ( X , y ) \mathcal{D} \equiv (X, y) D≡(X,y):其中包含12个核心特征( X = ( x 1 , . . . , x N ) X = (x_1, ..., x_N) X=(x1,...,xN), x n ∈ R D x_n \in \mathbb{R}^D xn∈RD, D = 12 D=12 D=12),以及对应的类别标签( y = ( y 1 , . . . , y N ) y = (y_1, ..., y_N) y=(y1,...,yN), y n ∈ { 0 , 1 } y_n \in \{0,1\} yn∈{0,1});下标(n)代表数据集中的不同材料(共 N N N个条目)。将分类任务转化为两个独立的回归任务后,我们基于共享核函数 K ( x , x ′ ) K(x, x') K(x,x′)搭建了两个高斯过程回归(GPR)模型。
核函数内部的相关矩阵(\mathbf{M})会耦合不同的核心特征,其设计采用因子分析结构 M = L L T + Λ \mathbf{M} = \mathbf{L}\mathbf{L}^T + \mathbf{\Lambda} M=LLT+Λ------这能让我们以有限的超参数学习不同核心特征(PF)间的相互作用。我们通过最大化对数边缘似然训练整个数据集的模型,这一过程会自动实现正则化。核心特征间的相关性 M ∗ M^* M∗可从训练后的模型参数中得到:第 m m m个与第 n n n个核心特征的耦合程度由标准化相关系数 C m n C_{mn} Cmn衡量,这能揭示识别方网拓扑半金属(TSM)的关键特征对。
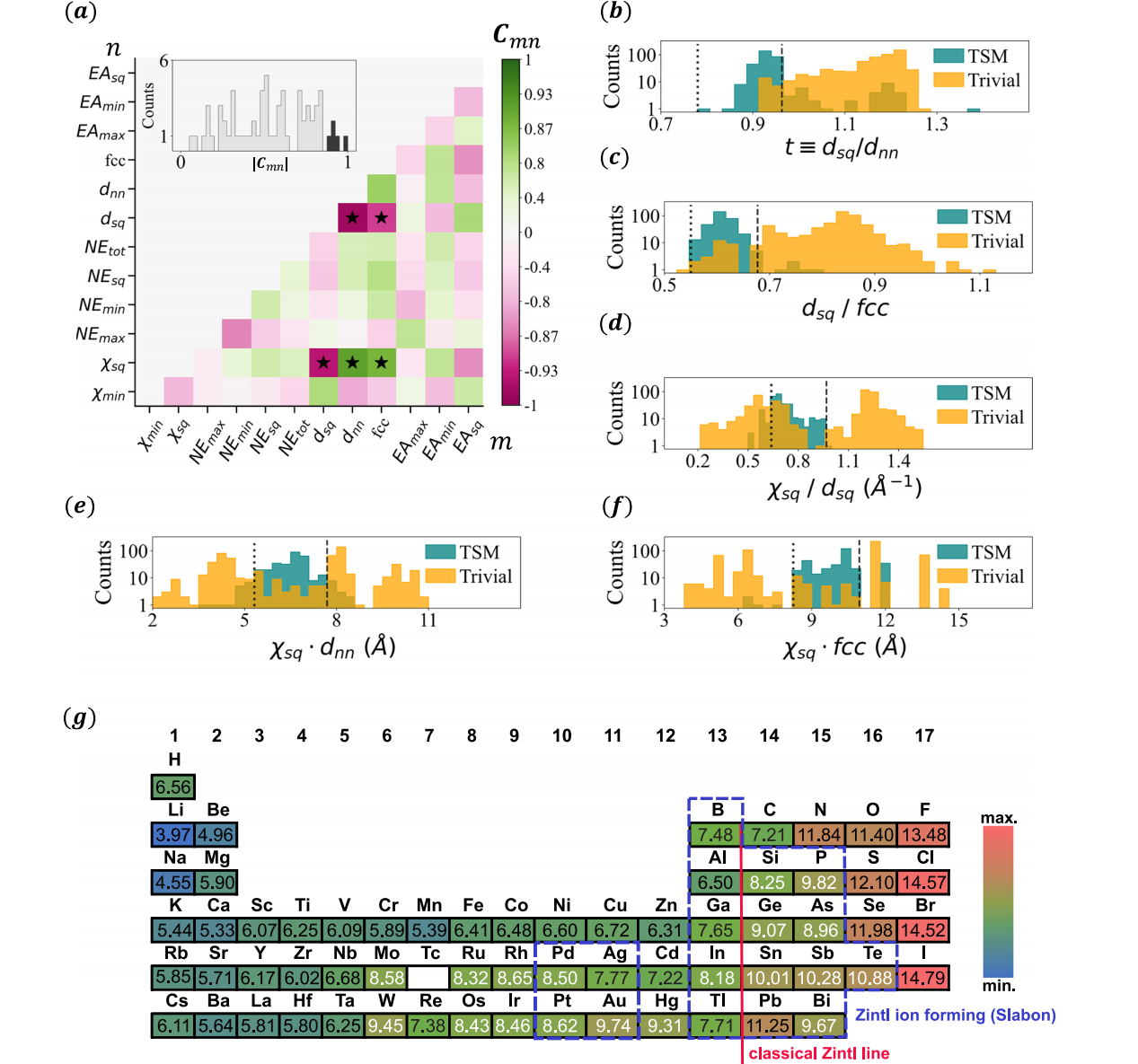
图3 | ME-AI的主要结果
图中展示了标准化相关矩阵 C m n C_{mn} Cmn( m m m、 n n n代表核心特征),仅呈现下三角部分;插图为非对角元素 ∣ C m n ∣ |C_{mn}| ∣Cmn∣的大小分布,黑色标注的是最强元素。关键特征对 ( d sq , d nn ) (d_{\text{sq}}, d_{\text{nn}}) (dsq,dnn)、 ( d sq , fcc ) (d_{\text{sq}}, \text{fcc}) (dsq,fcc)、 ( χ sq , d sq ) (\chi_{\text{sq}}, d_{\text{sq}}) (χsq,dsq)、 ( χ sq , d nn ) (\chi_{\text{sq}}, d_{\text{nn}}) (χsq,dnn)、 ( χ sq , fcc ) (\chi_{\text{sq}}, \text{fcc}) (χsq,fcc)用星号标记。
b d sq / d nn d_{\text{sq}}/d_{\text{nn}} dsq/dnn、c d sq / fcc d_{\text{sq}}/\text{fcc} dsq/fcc、d χ sq / d nn \chi_{\text{sq}}/d_{\text{nn}} χsq/dnn、e χ sq ⋅ d nn \chi_{\text{sq}} \cdot d_{\text{nn}} χsq⋅dnn、f χ sq ⋅ fcc \chi_{\text{sq}} \cdot \text{fcc} χsq⋅fcc在数据集中拓扑半金属(TSM)与平庸材料中的取值;竖线标记了支持拓扑半金属的新兴描述符的取值范围。
g 包含新元素特异性描述符 χ sq \chi_{\text{sq}} χsq的周期表:采用面心立方(fcc)配色(从最低值到最高值由蓝到红);面板f中,数值在8到11之间的条目(白字)是拓扑半金属的代表元素。红线代表经典津特耳线,蓝框元素是已知能形成津特耳离子的元素(如Slabon⁴⁴所述)。
📌 2 适用于元素周期表的通用图深度学习原子间势
A universal graph deep learning interatomic potential for the periodic table nature computational science
主要贡献
✅1)开发了一个通用的原子间势能函数(Universal IAP)------M3GNet
论文提出了一个基于图神经网络的通用原子间势函数模型 ,称为 M3GNet(Many‑body 3‑body Graph Network) ,它能够
✅ 覆盖周期表中绝大多数元素(89种元素)
✅ 对不同材料体系进行准确的能量、力和应力预测
该势函数可用于机器学习驱动的材料模拟,如晶体结构弛豫、分子动力学、热力学性质计算等。(Nature1)
✅ 2) 通用性:覆盖周期表中的绝大多数元素
M3GNet 是目前最全面的通用 IAP 之一,它不再只针对某一类材料,而是可以处理 89种元素组合与其化合物系统 。
这意味着可以用一个模型同时模拟金属、半导体、陶瓷等材料,而不需要为每种化学体系额外训练不同势函数。
✅ 3) 构建了高精度的图神经网络架构
论文的模型结合了
✔ 图神经网络(GNN)结构(节点代表原子,边代表邻接信息)
✔ 引入了 三体相互作用特征(many‑body/three‑body)
传统 GNN 只能表达 pairwise 信息,但三体信息让势能预测更接近真实物理行为(连续能量、力和应力)。(MatGL2)
✅ 4). 利用大规模 DFT 数据库作为训练集
模型训练使用了 Materials Project 提供的高通量 DFT 计算数据,包括:
📊 ~~187,000+ 个结构的能量
📊 ~16,000,000 种力
📊 ~1,600,000 种应力
这些大规模高质量数据是模型能够精准的关键基础。(materialsvirtuallab.org3)
✅ 5). 同时预测能量、力、应力,并能直接反向求力(自动求导)
通过对能量进行自回传自动求导(auto‑diff),模型可以:
✔ 算出各原子的力
✔ 得到应力张量
从而可以用于分子动力学与材料稳定性模拟,这在大多数 MLIP 中是很难兼顾的。(materialsvirtuallab.org3)
✅ 6). 大规模材料发现与筛选
利用这个势函数,作者对 ~31 M(百万)假设结构 进行了筛选,识别了 ~1.8 M 个潜在稳定材料 ,
并通过 DFT 验证了其中 约1578种稳定结构,展示了这套模型在大规模材料发现中的应用潜力。(arXiv4)
摘要
原子间势(IAP)用于描述原子的势能面,是原子尺度模拟的基础输入。然而,现有原子间势要么仅适配窄范围化学体系,要么在通用场景下精度不足。本文报道了一种基于图神经网络(M3GNet)的通用原子间势,可覆盖元素周期表中的多体相互作用。
M3GNet势以"材料项目"近十年积累的大规模结构弛豫数据集为训练基础,可广泛应用于不同化学空间材料的结构弛豫、动力学模拟与性质预测。研究从3100万种假设晶体结构中,筛选出约180万种基于M3GNet能量判定的潜在稳定结构;对能量最低的2000种结构,通过密度泛函理论验证后,1578种被确认为稳定。该结果证明:机器学习加速了"可合成材料"的发现路径,助力挖掘具备目标性质的新材料。
正文
原子尺度模拟是计算机辅助材料设计的基石。多数材料计算研究的第一步是获取平衡结构,这需要在势能面(PES)中搜索极小值------涉及遍历独立晶格、原子自由度,以寻找能量最小值。原子尺度模拟也被用于研究材料体系的动力学演化(如预测热力学平均性质),例如通过分子动力学模拟获得材料的热导率、熔点等性质¹。
尽管从头算方法(如密度泛函理论,DFT)能最精准地描述势能面,但其计算成本极高:不仅难以扩展到大规模材料研究,对体系尺寸的适配性也很差。因此,线性标度的原子间势(IAP) 常被用于描述势能面。这类势函数多以"对势"形式表示原子间相互作用,但多数只能适配单一元素,或至多覆盖2--5种元素的窄范围化学体系。
目前最流行的通用势场是AMBER力场²:它最初为分子/晶体体系设计,但主要支持分子模拟,在晶体体系中的精度与适用性有限。近年来,势能面的机器学习成为原子间势开发的极具前景的方法³⁻⁵。这类"机器学习原子间势(ML-IAP)"通常将势能面表示为局部环境描述符(如原子间距、键角、原子密度)的函数,已被证明在广泛化学体系中显著优于经典原子间势。
然而,此前的图深度学习模型⁶⁻⁸虽能实现高精度预测,却未验证其对元素周期表所有化学体系的普适性。因此,目前尚无适用于任意晶体结构的通用原子间势。
过去十年,高通量自动化框架⁹⁻¹⁰的出现,推动了可靠电子结构数据(包括"材料项目"¹¹、Open Quantum Materials Database¹²、Computational Materials Repository¹³等)的发展。这些数据库的核心是"结构弛豫计算":即通过电子结构计算得到平衡结构、能量、能带结构及衍生材料性质,用于材料筛选与设计。材料项目积累的海量"结构弛豫数据"(即中间结构及其对应能量、力与应力),为原子间势开发提供了基础。
方法与结果
本文提出一种图基深度学习原子间势 ,融合了传统原子间势的多体特征与灵活的图材料表示。利用"材料项目"自2011年成立以来积累的187000次弛豫、1600万条力、160万条应力的未开发数据集,我们训练了覆盖元素周期表89种元素的通用原子间势(M3GNet),可描述三体及多体相互作用。
M3GNet的应用场景包括声子与弹性计算、结构弛豫等;我们进一步弛豫了3000万种假设结构,以助力新材料发现。
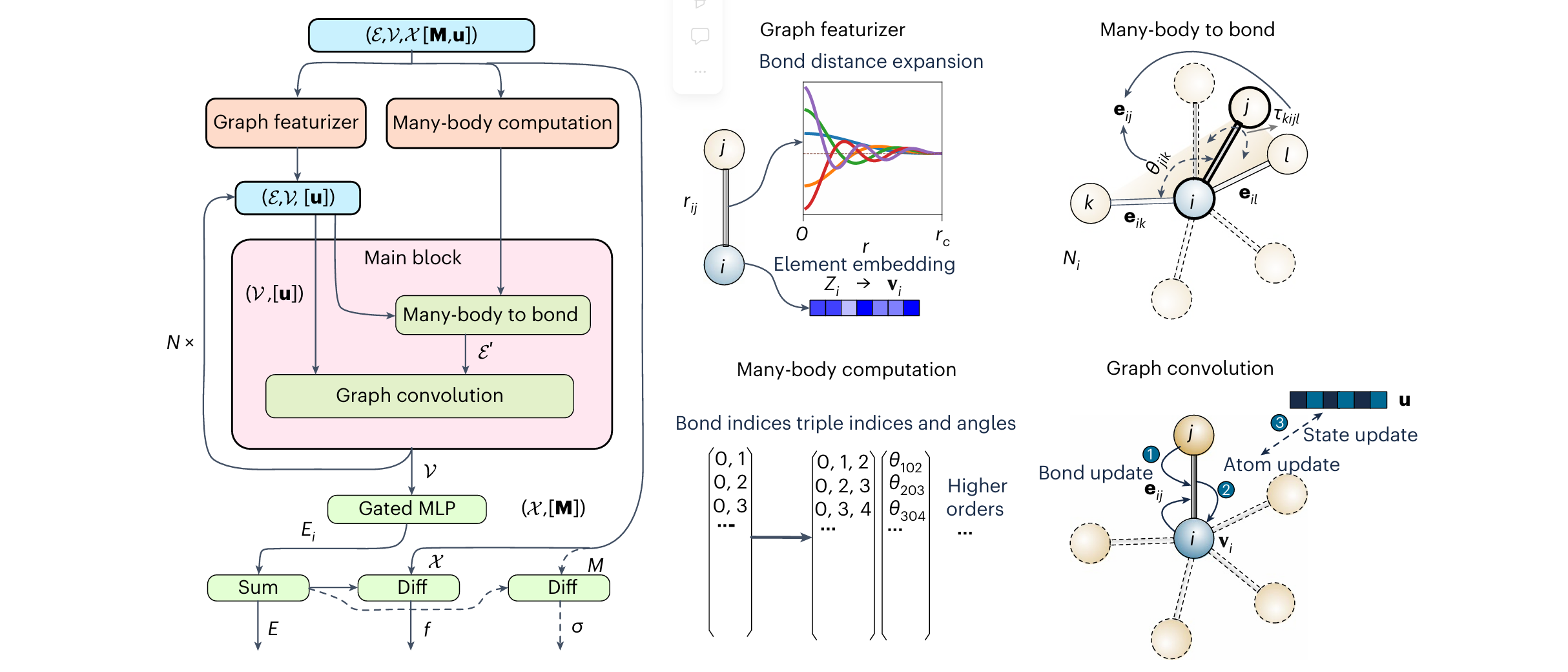
图1 多体图势与主要计算模块的示意图
模型架构始于包含原子与键的图,随后通过能量、力、应力输出的特征化与计算过程。主要模块包含图特征提取器与多体计算模块:
- 图特征提取器:将原子序数与元素嵌入可学习的连续特征空间;将键扩展为基函数,计算至多二阶的数值与导数(边界处设为0)。
- 多体计算模块:计算三体及多体相互作用的原子索引,以及原子间键的多体信息。
标准图卷积模块包含两个主要步骤:多体成键步骤与标准图卷积步骤。多体成键步骤通过考虑全键合环境 (如键角 θ i − j − l \theta_{i-j-l} θi−j−l、二面角 γ i − j − l − k \gamma_{i-j-l-k} γi−j−l−k等,以及键长 r i − j r_{i-j} ri−j等),计算原子 i i i的新键信息 Y i Y_i Yi;标准图卷积则迭代更新键与原子信息。
在聚合阶段,原子信息被传递至门控多层感知器(MLP)以获取原子能量,求和得到总能量;总能量的导数即为力与应力输出。
含多体相互作用的材料图
数学图是晶体与分子的天然表示方式:节点代表原子,边代表原子间的化学键²¹⁻²²。传统图神经网络模型在材料性质预测中表现优异²³⁻²⁴,但作为通用原子间势的效果有限------核心问题是缺乏"能量与力随键数变化的连续性"等物理约束。
本文开发了显式融合多体相互作用的材料图架构 :材料表示为 G = { V , E , T , U , M } \mathcal{G} = \{ \mathcal{V}, \mathcal{E}, \mathcal{T}, \mathbf{U}, \\mathbf{M} \} G={V,E,T,U,M},其中 V i ∈ V \mathcal{V}i \in \mathcal{V} Vi∈V是原子(i)的信息, E i − j ∈ E \mathcal{E}{i-j} \in \mathcal{E} Ei−j∈E是连接原子(i)与(j)的键的全局状态信息, T \mathcal{T} T是可选的键信息(如坐标), U \mathbf{U} U是单元信息, M \\mathbf{M} M是晶体的3×3平移矩阵。
图特征提取器将原子间距 r i − j r_{i-j} ri−j(至某截断值 r c r_c rc)嵌入基函数,并将原子序数 Z i Z_i Zi映射至元素特征空间。
模型开发借鉴了传统原子间势的思想,例如Tersoff键序势²⁵:通过原子 i i i的近邻 j j j、 l l l的所有键组合(排除 i i i和 j j j),纳入 n n n体相互作用。本文中,我们用图神经网络生成高阶相互作用(如键角、二面角),并将其与图相互作用聚合,随后将这些信息传递至标准图卷积步骤。
标准图卷积步骤可重复多次,以构建任意复杂度的图,类似此前的材料网络架构²³。本文仅聚焦三体相互作用的融合。
在原子间势拟合场景中,原子信息映射至原子能量 E i E_i Ei,求和得到总能量 E E E;总能量用于通过自动微分计算力与应力 σ \sigma σ。
M3GNet原子间势
基于M3GNet架构开发原子间势时,我们以晶体结构为输入,以对应的能量 E E E、力 f \mathbf{f} f、应力 σ \sigma σ为训练目标。模型通过自动微分生成力与应力: f = − ∂ E / ∂ x \mathbf{f} = -\partial \mathcal{E}/\partial \mathbf{x} f=−∂E/∂x( x \mathbf{x} x为原子坐标), σ = ∇ V ∂ E / ∂ ϵ \sigma = \nabla_V \partial \mathcal{E}/\partial \epsilon σ=∇V∂E/∂ϵ( V V V为体积, ϵ \epsilon ϵ为应变)。
原子间势数据集基准测试
我们选取了包含元素晶体(如面心立方镍、面心立方铜、体心立方锂、体心立方钼、金刚石硅、金刚石锗)的多样化密度泛函理论数据集,以评估M3GNet的性能。
从表1可见:M3GNet原子间势显著优于 嵌入原子法(EAM)、改进型嵌入原子法(MEAM)等经典多体势;其性能也与Behler--Parinello神经网络势(NNP)、矩张量势(MTP)等基于局部环境的机器学习原子间势相当。
需注意:尽管部分机器学习原子间势的能量与力误差略低于M3GNet,但它们在处理多元素化学体系时灵活性大幅下降------因为多元素适配会导致回归系数的组合爆炸,对数据量的需求也急剧增加。
相比之下,M3GNet架构将每个原子(节点)的元素信息表示为可学习的嵌入向量,因此能自然适配多元素化学体系。例如,针对6种元素训练的M3GNet-all势,性能与单独针对每种元素训练的M3GNet势相当。
与其他图神经网络模型类似,M3GNet无需增大键构造的截断半径即可捕捉长程相互作用(补充图1);同时,与此前的图神经网络模型不同,M3GNet架构能保持"能量、力、应力随键数变化的连续性"------这是原子间势的关键要求(补充图2)。
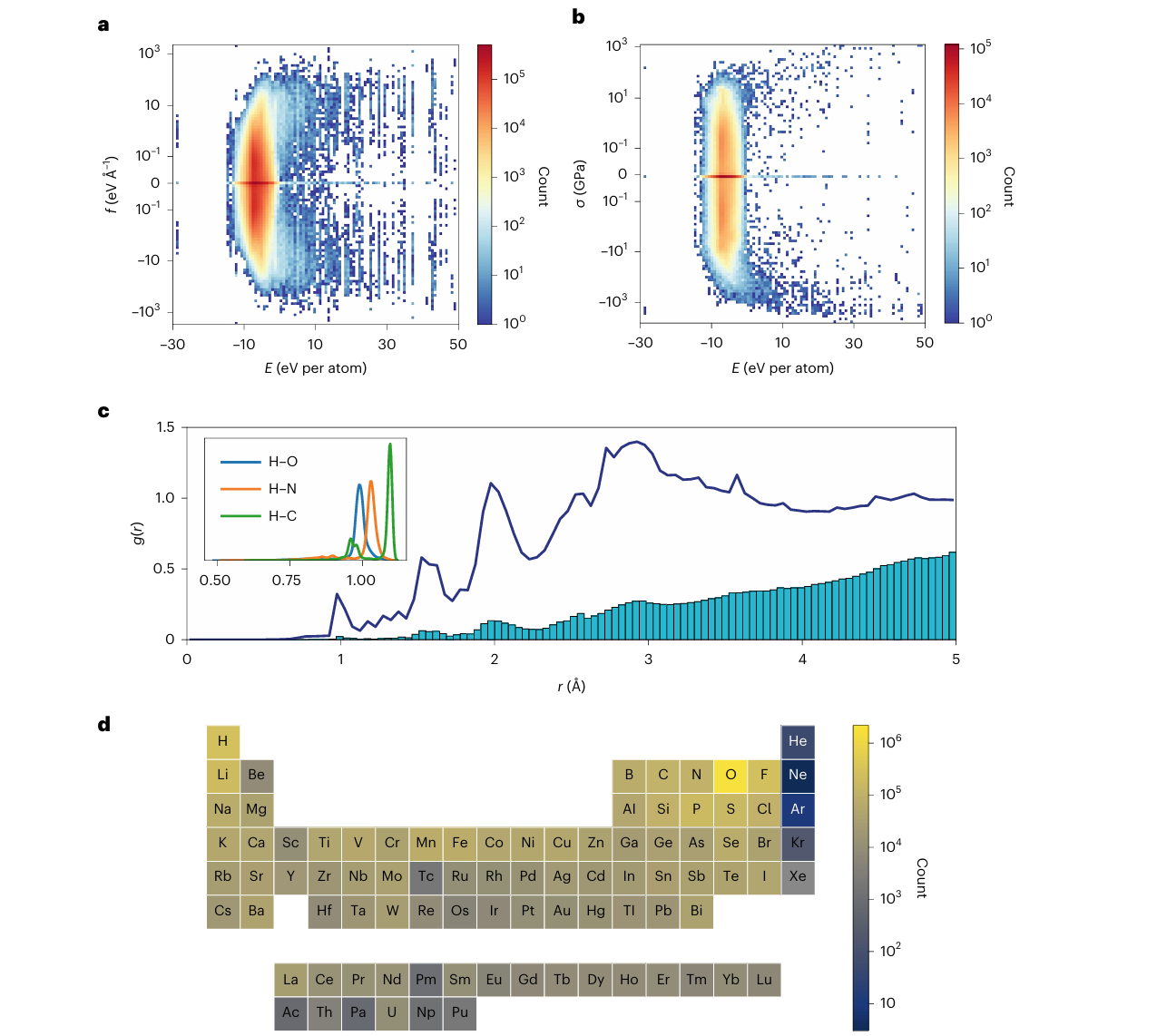
图2 | MPF.2021.2.8数据集的分布
a、b,单原子结构能( E E E)与力(a)、应力(b)分量的分布关系。
c,径向分布函数 g ( r ) g(r) g(r)(深蓝色线)与原子对间距分布密度(浅蓝色直方图);短间距(<1.1 Å)的密度由主要与氧、碳、氮形成氢键的原子构成,如插图所示。
d,数据集中所有原子的元素计数,覆盖了元素周期表中的89种元素。
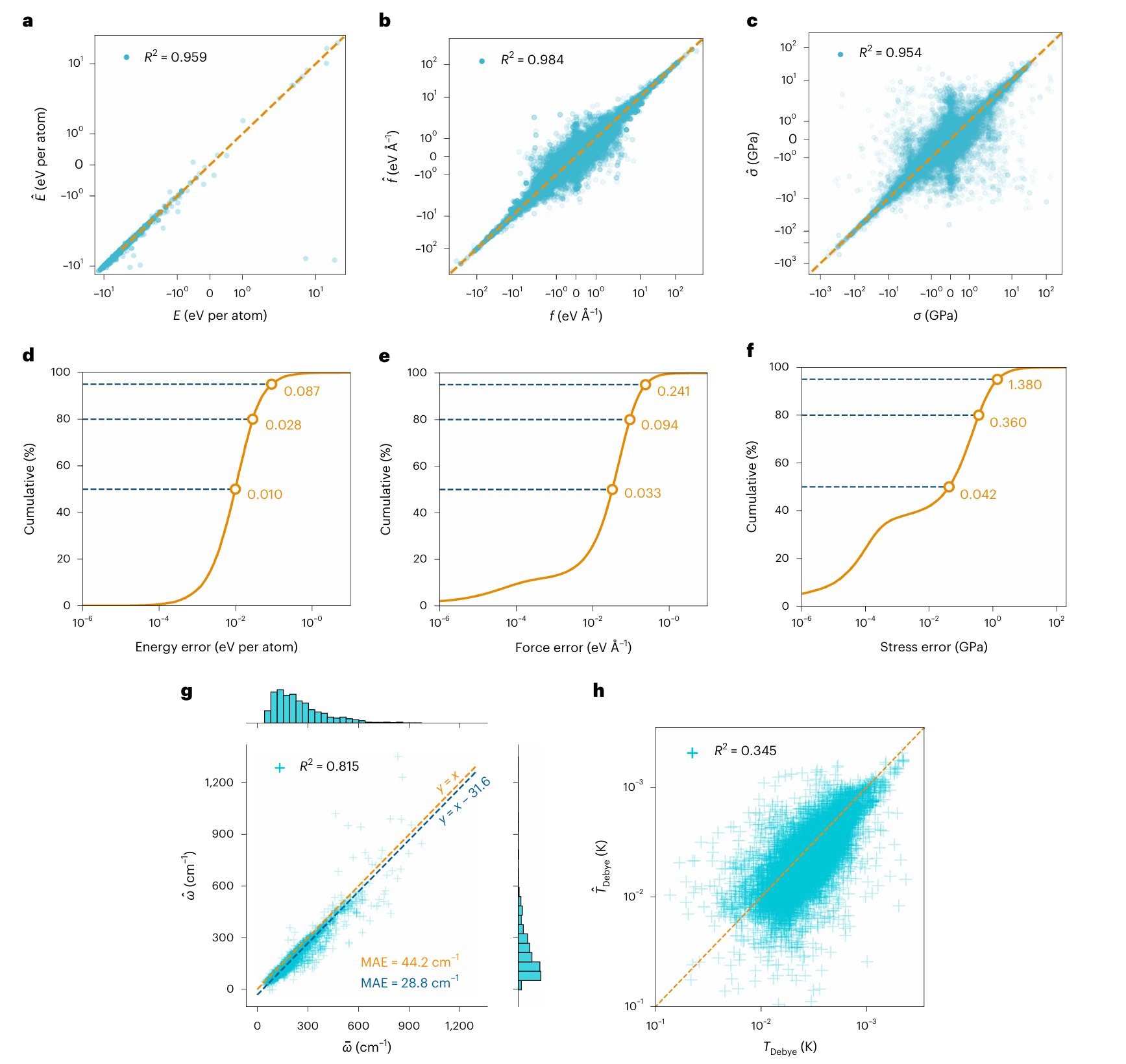
图3 | 测试数据集上的模型预测结果与DFT计算的对比
a--c,能量( a a a)、力( b b b)和应力( c c c)的一致性图。模型预测结果记为 E ^ \hat{E} E^、 f ^ \hat{\mathbf{f}} f^和 σ ^ \hat{\boxed{\sigma}} σ^;虚线( y = x y = x y=x)为参考线。
d--f,能量( d d d)、力( e e e)和应力( f f f)的误差累积分布;水平虚线分别对应模型在50%、80%和95%处的误差(从下到上)。
g,模型计算的1521个声子态密度中心数据( ω ^ \hat{\omega} ω^),与Petretto及其合作者²⁹的PBEsol DFT计算结果( ω \omega ω)的对比。
h,由M3GNet模型得到的11848个德拜温度(排除负模量)( Θ ^ Debye \hat{\Theta}_{\text{Debye}} Θ^Debye),与de Jong等人⁶⁴基于PBE DFT弹性张量的计算结果的对比。
📌3.利用少样本机器学习实现电子显微镜数据的快速灵活分割
Rapid and flexible segmentation of electron microscopy data using few-shot machine learning npj computational materials
这篇论文 "Rapid and flexible segmentation of electron microscopy data using few‑shot machine learning" (发表在 npj Computational Materials 2021)提出了一种能够在仅需极少标注样本(few‑shot)的情况下,对扫描透射电子显微镜(STEM)图像 进行快速且灵活的语义分割的方法,解决了传统显微图像分析难以泛化和高成本标注的问题。(Nature1)
✅ 1) 机器学习用于 STEM 图像分割
论文提出了一个 few‑shot(少样本)机器学习框架 ,用于对 STEM(扫描透射电子显微镜)图像进行 语义分割(semantic segmentation),即将图像中的不同微观区域(例如界面、相区、纳米粒子、基底背景等)自动标注出来。(Nature1)
传统分割方法(如阈值、清洗、聚类等)
❌ 通常对噪声非常敏感
❌ 难以跨不同材料类型泛化
❌ 需要大量手工调整参数/标注数据
而本文提出的 few‑shot 方法能以 极少的标注示例(5 -- 8 个小图像片段) 即可对整张高分辨显微图像进行有效分割,大幅减少了标注工作量。(Nature1)
✅ 2)少样本学习(Few‑Shot Learning)基本思路
- 传统深度学习分割需要成千上万带标签的训练样本;
- Few‑shot 学习则旨在仅用少量"support set"(支持集)示例学习新类别,从而对新图像进行分类/分割。(Nature1)
✅ 3)具体做法
-
划分 Chips(小图像块)
将高分辨率的 STEM 图像切成许多小的"chip"(子图像);
-
构建支持集(Support Set)
从这些 chips 中,手工标注极少的示例作为每个类别的"原型(prototype)";
-
利用原型网络(Prototypical Network)分类
- 每个类别的 support chips 都被映射到一个向量空间(用深度神经网络 Embedding);
- 利用这些支持集的 embed 向量构造每个类别的 prototype;
- 将待分类的 query chip 和各类别 prototype 计算距离,进行分类;
-
重构分割图
将分类结果拼接成最终分割图。(Nature2)
这种方法本质上与一般的分类任务类似,但它依赖于"距离度量而非大规模训练集",从少数示例即可学习出特征识别能力。(Nature2)
✅ 4)实验与表现
论文在三类典型材料的 STEM 图像上进行了验证:
- SrTiO₃/Ge 外延异质结构
- La₀.₈Sr₀.₂FeO₃ (LSFO) 薄膜
- MoO₃ 纳米粒子
这些材料具有不同的微结构特征、形态、尺度和噪声分布。few‑shot 方法在以上数据上均展示了良好的分割效果,而且对噪声和不同对比度的图像较传统方法鲁棒性更强。(osti.gov3)。 这篇论文提出了一种基于 few‑shot machine learning 的显微图像分割方法,通过少量样例就能对复杂的 STEM 显微图像进行快速且鲁棒的语义分割,是推动高通量、智能材料表征的重要工作。
摘要
原子尺度电子显微镜图像中关键微观结构特征的自动分割,对加深理解众多重要材料、化学体系的结构-性能关系至关重要。但当前方法依赖耗时的人工分析------这类分析本质上基于经验、易出错,既无法处理现代仪器产生的海量多样化数据,也难以泛化到不同的微观结构特征与材料体系。
本文提出一种灵活的半监督少样本机器学习方法,用于扫描透射电子显微镜(STEM)图像的分割。我们以三种氧化物材料体系为验证案例:(1) S r T i O 3 ( S T O ) SrTiO₃(STO) SrTiO3(STO)/ G e Ge Ge的外延异质结构、(2) L a 0 . 8 S r 0 . 2 F e O 3 La₀.₈Sr₀.₂FeO₃ La0.8Sr0.2FeO3( L S F O LSFO LSFO)薄膜、(3) M o O 3 MoO₃ MoO3纳米颗粒。结果表明:该少样本方法比现有方法更稳健、对噪声更不敏感,且所需的微观结构特征标注更少。此方法可实现快速图像分类,为高通量表征与自主显微镜平台赋能。
引言
材料的微观结构决定了催化剂、储能器件、量子计算等众多重要技术的功能。扫描透射电子显微镜(STEM)长期以来是研究微观结构的基础工具------它能以原子尺度分辨率解析结构、化学组成与缺陷,可表征各类材料¹⁻³。
传统上,STEM图像需人工或半自动分析:由专家先验知识指导,逐像素分类以解析不同微观结构特征⁴⁻⁵。这种方法适用于小数据量,但面对现代实验产生的海量、高噪样本时并不实用⁶⁻⁷;同时难以快速扩展,阻碍多模态数据的高通量分析,也无法与现代仪器协同⁸。更根本的是,这类方法未充分发挥测量潜力,还影响实验可重复性⁹,在复杂合金(性能受大量未知缺陷影响¹⁰⁻¹²)的分析中局限尤为突出。因此,亟需更快速、更严谨的微观结构表征方法。
定量描述显微镜图像的核心挑战,是微观结构与数据模态的多样性:同一台仪器可能先分析原子尺度界面,下一次就研究纳米颗粒或晶粒边界。在扫描电子显微镜(SEM)中,需将语义模糊的模态描述符与物理模型关联,这让精确量化特定STEM的物理意义变得困难¹³⁻¹⁴。例如,通过图像分割估算特定相的面积分数,是建立结构-性能关系的关键环节¹⁵⁻¹⁶。
尽管大津阈值法¹⁷、分水岭算法¹⁸等分割方法已被应用,但它们多针对特定体系定制,不易泛化且需大量预处理²⁰。机器学习(尤其是卷积神经网络)虽已用于微观结构识别²¹⁻²⁶,但传统深度学习需海量标注数据³³⁻³⁵,而显微镜研究中常仅能获得少量训练样本³⁸⁻³⁹。
近期研究表明,轻量级学习技术仅需少量标注即可实现自动分割³⁶·³⁷,但材料科学领域对这类方法的探索有限³⁶。少样本/单样本学习还适用于瞬态/不稳定材料(样本有限或实验周期长)的研究,也可用于分析有限的历史数据⁴³。
本文提出一种少样本机器学习方法,用于STEM图像的快速分割。我们选择三种氧化物体系(STO/Ge异质结构、LSFO薄膜、MoO₃纳米颗粒)进行验证------它们具有丰富的微观结构特征,且在半导体、自旋电子学等领域有重要应用⁴⁴⁻⁴⁵。
结果显示:仅需5--8个"子图像芯片"(特定微观结构特征的示例),模型即可得到与领域专家相当的分割结果。该方法噪声敏感性低、学习能力强,能快速识别STEM数据流中的微观结构,为实时分析提供支持,也凸显了图像驱动机器学习在高通量表征中的潜力。
结果与讨论
我们开发了一种名为"少样本学习"的深度学习方法,用于扫描透射电子显微镜(STEM)图像的超像素分割 (即对STEM图像中的超像素进行分类)。少样本学习的核心思路是:仅用少量(<10个)已标注的目标区域示例,让模型识别图像中对应类别的区域。这种半监督方法能利用材料微观结构中常见的重复模式,无需大量标注即可训练模型------甚至仅用单个标注示例,就能完成图像分割。
少样本学习实现图像分割的通用流程是:先将图像拆分为若干子图像(称为"芯片"),经模型初始化、推理后输出分割后的显微图(图1)。该方法依赖计算机视觉领域的域特定知识(如图1所示的标注技术),这类技术通常被称为"语义分割"------相比像素分类,语义分割的分类粒度更精细,但在噪声图像或像素值波动较大的场景中易出错。而少样本分割方法通过捕捉芯片中的特定微观结构特征,能更好地呈现细节;针对STEM图像的少样本纹理分割研究,也为改进微观结构表征、助力材料发现提供了可行路径。
预处理
为分离并测量STEM图像中对比度变化的不同物相,需对原始图像数据进行预处理。我们选用"限制对比度自适应直方图均衡化(CLAHE)"技术³⁹,用于增强局部图像质量;其实现细节见表1。预处理步骤为:先对原始图像执行CLAHE处理,再将处理后的图像分割为更小的子图像(图1b)。芯片尺寸在95×95像素到32×32像素之间调整------尺寸需足够大以捕捉可变结构,同时足够小以适配嵌入模块。
预处理的第二步是增强相邻空间区域的粒度(图1),通过"高斯LOG斑点检测算法"³⁹实现(该技术可同时测量特征的位置与尺寸)。我们在La₀.₈Sr₀.₂FeO₃(LSFO)体系中应用了这一步骤,以增强极微弱的相差异。
少样本模型
少样本模型输入预处理后的STEM图像(通常为高分辨率图像,约3000×3000像素),并将其拆分为若干更小的芯片(一般不超过100×100像素)。每个芯片可作为示例,或用于定义"支持集"以描述一个/多个类别。多数少样本应用会为每个类别创建独立的支持集⁵²⁻⁵⁴,因此我们将原始图像拆分为更小的子图像网格(图1b),选取其中一个子集标注为单类别,从而为每个类别定义支持集。
支持集 i i i由 N N N个标注芯片 X i = ( x i , 1 , . . . , x i , N ) X_i = (x_{i,1}, ..., x_{i,N}) Xi=(xi,1,...,xi,N)及对应"真实"类别 Y i = ( y i , 1 , . . . , y i , N ) Y_i = (y_{i,1}, ..., y_{i,N}) Yi=(yi,1,...,yi,N)组成(图1c)。
本文选择原型网络 作为相似性模块------因其轻量化设计与简洁性,可将每个支持集芯片 x i , j x_{i,j} xi,j表示为单个原型 c k c_k ck。模型通过嵌入函数 将每个芯片 x i , j x_{i,j} xi,j转换为 M M M维表示 z i , j = f θ ( x i , j ) z_{i,j} = f_\theta(x_{i,j}) zi,j=fθ(xi,j);随后,类别 k k k的原型 c k c_k ck定义为该类别支持集嵌入表示的均值向量,公式如下:
c k = 1 N k ∑ ( x i , j , y i , j ) ∈ S k z i , j (1) c_k = \frac{1}{N_k} \sum_{(x_{i,j}, y_{i,j}) \in S_k} z_{i,j} \tag{1} ck=Nk1(xi,j,yi,j)∈Sk∑zi,j(1)
原型构建完成后,未训练的原型网络会通过以下步骤对新的查询数据进行分类:先通过嵌入函数得到查询芯片的嵌入表示,再计算其与各原型的欧氏距离,最后通过softmax归一化得到类别概率------概率最高的类别即为查询芯片的标签(图1d)。模型的最终输出是每个查询芯片对应的类别标签(图1e)。
模型推理
为了量化STEM图像中的物相分数(空间维度可达数纳米至数微米),每个芯片都会作为查询样本:嵌入后计算与各原型的距离,得到类别分布,进而分配类别。需注意:若存在未知特征,建议在支持集中新增对应的类别。
模型实现与参数
模型的具体实现与参数详见表2。尽管模型参数选择通常较为繁琐,但少样本场景下的参数设置相对简单------因为可以利用预训练的嵌入架构。本文选用**101层残差网络(ResNet101)**⁵⁸作为嵌入架构,原因是其在多个相关图像识别任务中表现优异。也可采用其他编码网络,例如更轻量化的MS-DNets³⁷、U-Nets⁵⁹,或高度专用的DefectSegNet³¹。
ResNet是一种常用模型,在多种编程语言中均可获取,且预训练权重易于迁移至STEM特定任务;ResNet101的权重来自PyTorch⁶⁰(vision v0.6.0版本),基于ImageNet⁶¹数据集训练。需说明的是:针对显微镜图像优化的网络可能提升训练与推理性能,还能支持更大批次以增强正则化。此外,我们选用欧氏距离作为度量------该度量在各类基准数据集与分类任务中普遍表现良好⁵⁷。
相似性模块也可采用其他少样本/元学习架构,但原型网络通常更简单易实现。芯片尺寸、批次大小等非模型专属参数,需结合显微图尺寸与计算内存容量确定:芯片应包含单个显微结构,具体尺寸需根据全图大小与放大倍数调试;批次大小即一次评估的芯片数量。一般来说,配备至少16GB内存、2.7GHz处理器的机器,处理64×64像素的芯片时,每0.5秒可完成1个芯片的预测,批次大小为100。计算时间还与芯片尺寸、嵌入模块的参数数量相关;若要训练少样本模型(而非本文仅讨论的推理),则至少需要1块GPU,且在大型数据集(如含1400万张图像的ImageNet⁶¹)上可能需要数天才能收敛。
分类
采用原型架构的少样本分类,在三种氧化物体系中的分割结果如图2所示。模型输出为超像素分类(即同一芯片内的所有像素共用一个标签与对应颜色),这与其他计算机视觉应用的分割方式一致⁶³。支持集类别定义了所有可能的输出类别;图2右侧展示了各支持集类别的芯片占比,通过像素尺度转换可得到每个显微图中各类别的面积占比。
STO/Ge体系对多数图像分析技术而言是个挑战------其对比度在全图范围内不规则变化,该样本是典型的薄膜-衬底界面成像数据。所选LSFO图像中,钙钛矿结构基体中存在第二相,且第二相从上到下呈梯度分布,大幅缩小了两种显微结构的细微差异;而分离这两种交织的显微结构,是理解合成过程与电导率等性能的关键。
尽管预处理可调整部分不规则性,但大津阈值法²⁰、分水岭算法²¹等传统阈值分割技术无法提供稳定结果,自适应方法在多图泛化时也会失效。另一种物相发现方法(基于滑动FFT)¹⁸虽有效,但要求图像具有周期性,且不适用于图2c所示的低倍图像------此时对比度、边缘与采样参数的卷积会增加直接解释的难度;此外,该方法也不适合分类非晶材料(非晶材料的关联序无周期性,难以局部量化)。
少样本技术也并非完全不受这些不规则性的影响(如图2a中分割后的显微图存在少量错分芯片),这些错分反映了模型对支持集选择的敏感性,这是后续研究的重要课题。总体而言,基于规范特征(分辨率高、均匀)支持集的分类,性能优于基于异常特征(含噪声、不完整)支持集的分类。
在LSFO体系中,少样本方法识别(绿色)显微结构的结果略不稳定(图2b),但这些问题可通过后处理修正(例如:对某一半径内被单一标签完全包围的芯片,调整其类别概率,或修改定义支持集的芯片)。尽管存在少量不规则性,少样本方法相比其他分割技术,对全输入图像中的噪声具有更强的鲁棒性。
此外,该少样本方法易于泛化到多种材料体系:由单张图像定义的支持集,无需调整即可应用于同类型的多张图像,大幅节省了图像序列分析的时间。这一特性在大面积映射中尤为重要(如MoO₃纳米颗粒案例)------此类场景需要采集图像拼接图,以观测各类可能的颗粒形态。
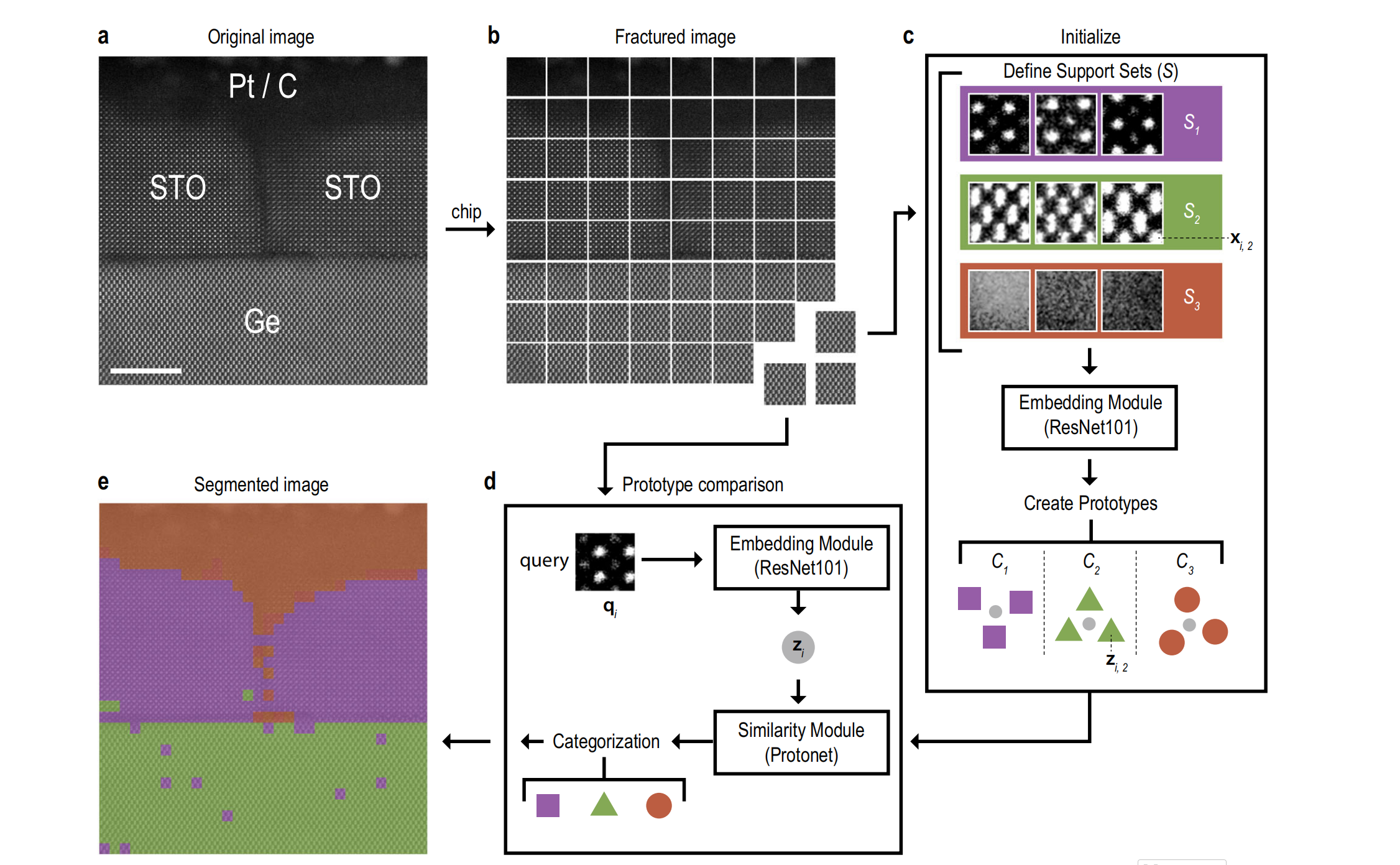
图1 少样本模型架构
原始STO/Ge图像(a)被拆分为若干子图像芯片(b);选取少量芯片在"支持集"中代表目标分割类别(c);每个芯片作为查询样本,与支持集定义的"原型"(d)通过欧氏距离对比分类,最终得到分割图像(e)。比例尺:5 nm。

图2 少样本分割结果
a--c 对三种材料体系(STO/Ge、LSFO、MoO₃)的分析:依次展示了原始图像(左)、对应的支持集(中),以及少样本分割的输出结果(右);每个体系中各支持集类别的占比估算值,呈现于对应子图的插图条形图中。比例尺: a -- b a--b a--b为5 nm, c c c为5 μm。