
研究论文
● 期刊: Nature Microbiology (IF 19.4)
● 原文链接: https://www.nature.com/articles/s41564-025-01983-z
● DOI: https://doi.org/10.1038/s41564-025-02042-3
● 第一作者:Younhun Kim
● 通讯作者:Bonnie Berge(bab@mit.edu),Travis E. Gibson(tegibson@bwh.harvard.edu)
● 主要单位:麻省理工学院、布莱根妇女医院、哈佛大学布罗德研究所、哈佛医学院、代尔夫特理工大学、华盛顿大学
摘 要
从宏基因组数据中随时间检测和定量微生物群落的能力在临床、基础科学和公共卫生方面有着广泛的应用。鉴于这些应用,以及病原体和其他感兴趣的分类单元可能以低相对丰度存在这一事实,迫切需要能够准确地以菌株水平分辨率对低丰度微生物分类单元进行分析的算法。在此,我们介绍ChronoStrain:一种用于纵向样本中菌株分析的序列质量感知和时间感知的贝叶斯模型。ChronoStrain明确地模拟每种菌株的存在或缺失,并为每种菌株的丰度轨迹产生一个概率分布。通过使用合成和半合成数据,我们展示了ChronoStrain在丰度估计和存在/缺失预测方面优于现有方法。将ChronoStrain应用于两个人类微生物组数据集,证明了其在分析成年女性反复泌尿道感染的纵向粪便样本中大肠杆菌菌株爆发以及在婴儿粪便样本中检测粪肠球菌菌株方面的改进解释性和准确性。与最先进的方法相比,ChronoStrain检测低丰度分类单元的能力尤为突出。
结果
ChronoStrain 概述
ChronoStrain 的流程如图 1 所示。在初始的生物信息学步骤中处理三个组件(图 1a,蓝色阴影框):
(1)实验的原始 FASTQ 文件
(2)基因组组装数据库
(3)标记序列"种子"数据库
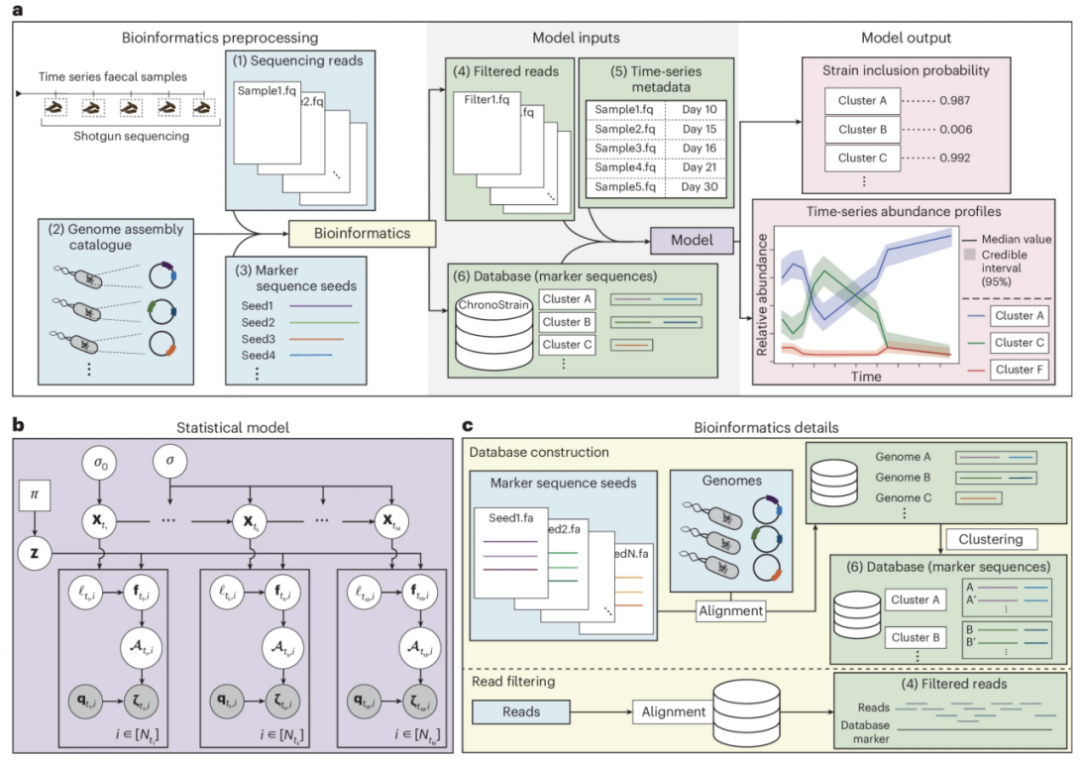
图1. ChronoStrain 概述
a,ChronoStrain 分析管道的高级示意图,显示了生物信息学预处理步骤的输入(蓝色)、模型输入(绿色)和模型输出(粉红色)。b,ChronoStrain 使用的概率模型(方法"潜在丰度模型"、"测序片段模型"和"测序噪声模型")的图形表示。白色圆圈是潜在随机变量,灰色圆圈是观测值,正方形是超参数(并非显示所有模型参数)。c,生物信息学预处理步骤的详细示意图,说明如何使用标记序列种子和参考基因组来构建菌株(簇)数据库,以及初始读取过滤过程的附加说明。
在初始的生物信息学处理过程中,执行了两项任务。首先,利用组件(2)和(3)为将要分析的每种菌株生成一个定制的标记序列数据库(方法------"ChronoStrain数据库")。然后,将(1)中的原始序列读取数据与该数据库进行过滤,得到一组过滤后的序列读取数据(方法------"序列读取过滤")。ChronoStrain贝叶斯模型的输入如下(图1a,绿色阴影框):
(4)带有质量分数的过滤后的序列读取文件(FASTQ格式)
(5)包含样本时间点信息的元数据文件
(6)为每种被分析的菌株定制的标记序列数据库
模型的两个最有用的输出是样本中每种菌株存在/缺失的概率,以及数据库中每种菌株的(概率性)时间序列丰度曲线(图1a,粉色阴影框)。我们现在更详细地讨论流程和模型的核心组成部分。
在本研究中,我们对菌株的定义是一个操作性的定义:菌株仅仅是标记序列的集合。用户不是手动为每个基因组指定标记,而是指定标记序列"种子"。这些种子不必是基因本身;它们可以是任意的核苷酸序列。在本研究中发现的候选标记种子包括MetaPhlAn核心标记基因、序列分型基因、菌毛基因以及其他已知的致病因子。每个种子都与参考数据库基因组进行比对,每个足够相似的匹配都被识别为数据库中相应基因组的标记序列。
作为最后一步,用户可以决定是否对参考序列进行聚类以及聚类的阈值,从而选择区分不同菌株的粒度。在本研究中,我们使用不同的阈值进行菌株聚类,范围从99.8%的序列相似性到约100%(每种独特的标记序列组合都被视为不同的菌株)。我们再次强调,"菌株簇"和"菌株"是互换使用的。
我们的贝叶斯模型(用于单个时间序列)如图所示。1b. 应变丰度使用随机过程建模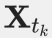 (由每个菌株 s 索引为
(由每个菌株 s 索引为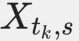 跨时间点 tk∈ {t1, ..., tM},以及模型包含变量 Z(索引为 Zs). 然后,在每个时间点 tk,第 i个读数被建模为核苷酸序列
跨时间点 tk∈ {t1, ..., tM},以及模型包含变量 Z(索引为 Zs). 然后,在每个时间点 tk,第 i个读数被建模为核苷酸序列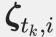 及其相应的质量得分向量
及其相应的质量得分向量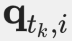 。序列
。序列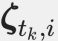 通过变量建模
通过变量建模 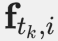 (读取的源核苷酸序列片段),
(读取的源核苷酸序列片段),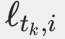 (沿标记的滑动窗口的随机长度,用于确定测量哪个片段)和
(沿标记的滑动窗口的随机长度,用于确定测量哪个片段)和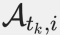 (片段读取替换/插入缺失误差曲线)。我们的贝叶斯推断的输出不是一个丰度估计;它实际上是一个完整的概率分布。然后可以直接询问这些分布以评估模型的不确定性。该模型的完整描述可以在方法中找到------"潜在丰度模型"、"测序片段模型"和"测序噪声模型"。
(片段读取替换/插入缺失误差曲线)。我们的贝叶斯推断的输出不是一个丰度估计;它实际上是一个完整的概率分布。然后可以直接询问这些分布以评估模型的不确定性。该模型的完整描述可以在方法中找到------"潜在丰度模型"、"测序片段模型"和"测序噪声模型"。
ChronoStrain 在基准测试方面优于其他方法
我们根据合成和半合成数据对 ChronoStrain 进行了基准测试。合成基准基于 CAMI2"应变疯狂"挑战。我们的半合成基准测试结合了 UMB 研究参与者的真实读数使用合成计算机读取。我们首先介绍半合成基准测试,然后以基于 CAMI2 的基准测试结束。
我们的半合成数据生成过程在图 2a 中概述。2a 中提供了更多详细信息,在"半合成数据生成"方法中提供了更多详细信息。真实读数取自 UMB 参与者 18 (UMB18) 的前六个纵向粪便样本,其中仅检测到系统群 B2 和 D 大肠杆菌菌株。合成读数由六个系统群 A 菌株生成,这些菌株经过合成突变,与参考数据库中的基因组不同。然后,使用预定义的时间(地面实况)丰度曲线,从六个突变菌株生成合成读数,然后与真实读数相结合。这些组合的读数集是现实的,同时具有用于评估的明确定义的地面实况丰度比概念。
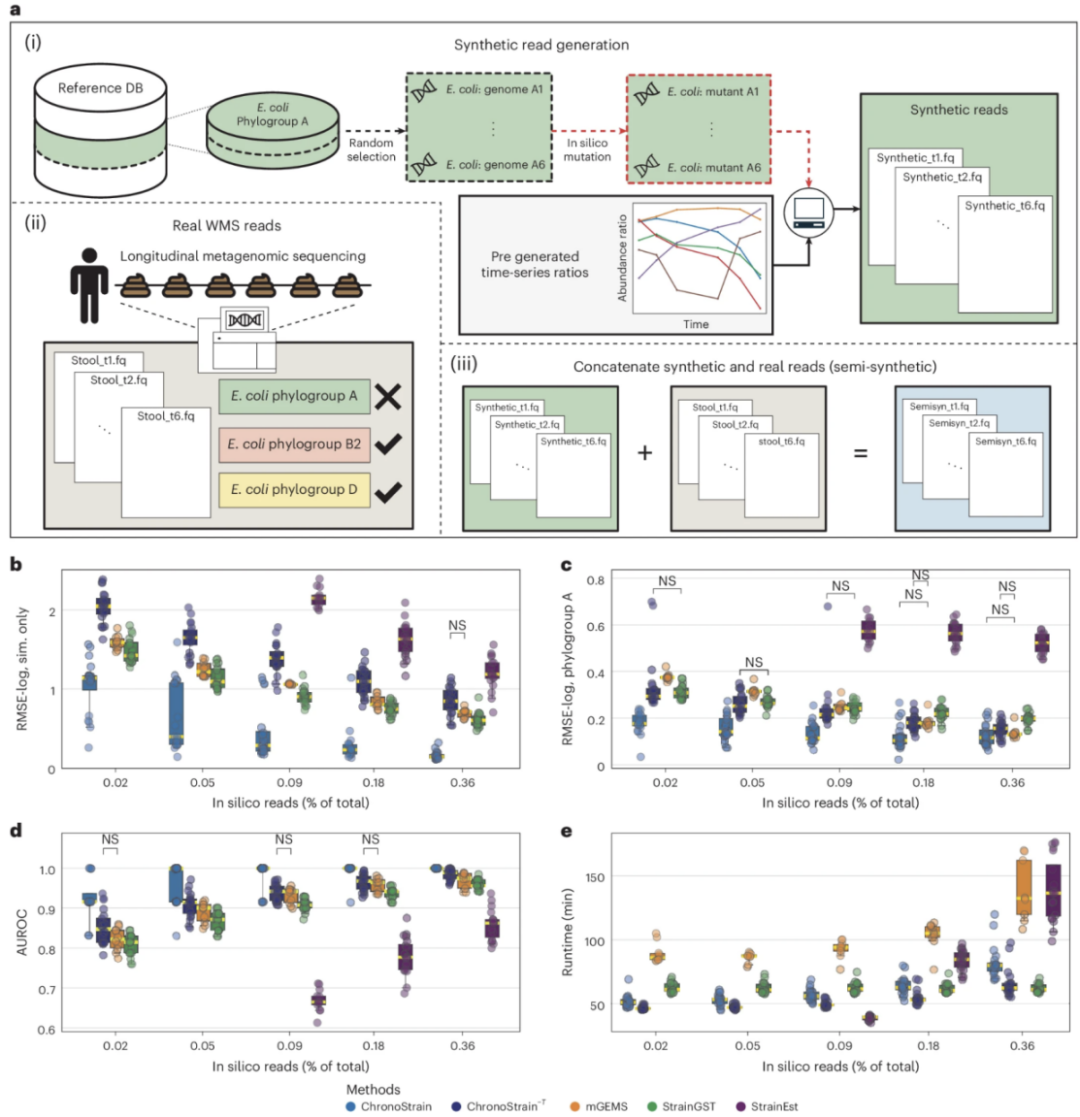
图2. ChronoStrain在半合成数据上的表现优于其他最先进的方法
a, 半合成数据生成过程的概述:(i) 从6个突变的A生态型大肠杆菌基因组中生成合成序列读取数据;(ii) 然后从参与者UMB18的前六个纵向样本中提取真实序列读取数据,这些样本仅鉴定出B2和D生态型的大肠杆菌;(iii) 最后,将真实和合成的序列读取数据进行拼接。b, c, 模型对数预测的均方根误差(RMSE)平均值在六个合成菌株上进行计算(b),并在数据库中所有A生态型菌株上进行评估(c),以考虑每种方法的估计值中包括假阳性的情况。d, 合成菌株检测的曲线下面积(AUROC)按A生态型进行标准化。e, 算法运行时间。每个测序深度有n=20个重复。除非特别注明NS(P值见补充表1),否则所有与ChronoStrain的比较在经过配对双侧Wilcoxon检验并进行Benjamini--Hochberg(BH)校正后,统计学上显著性水平为0.05。中位数用黄色表示,箱体为25%和75%分位数,须线为2.5%和97.5%分位数。
为了进行比较,我们纳入了StrainGST、StrainEst和mGEMS流程。关于未纳入基准测试的方法的讨论,请参阅补充文本A。在所有半合成基准测试中,我们以两种不同的模式运行ChronoStrain:时间序列感知模式和时间序列不可知模式(ChronoStrain−T),后者指的是独立地在每个样本上运行ChronoStrain。
在所有模拟的测序深度下,ChronoStrain在对数丰度的均方根误差(RMSE-log)和接收者操作特征曲线下面积(AUROC)方面显著优于所有其他方法,只有一个场景除外,同时保持与其他方法相当的运行时间(图2)。正如预期的那样,ChronoStrain−T的表现不如ChronoStrain,但在AUROC(图2d)和A生态型的RMSE-log(图2c)方面,它仍然显著优于或与其它比较方法相当。尽管ChronoStrain−T没有编码样本时间点信息,但它仍然明确地通过指示变量Zs对每种菌株的存在/缺失进行建模,这有助于控制假阳性。
当RMSE-log仅在六个目标菌株上计算时(图2b),方法不会因假阳性而受到惩罚,此时ChronoStrain−T的表现显著不如mGEMS和StrainGST。ChronoStrain在完整时间序列运行中的性能提升来自于其对低丰度菌株的更准确估计,这在我们将RMSE-log贡献根据合成菌株的样本丰度进行分箱时尤为明显(扩展数据图1)。我们还进行了正式的敏感性分析,调整了模型的超参数和真实基因组的突变率,相关内容可在补充文本C.2中找到。
我们的全合成基准测试基于"微生物组解释关键评估II"(CAMI2)的"菌株疯狂"挑战。原始的"菌株疯狂"挑战从408个基因组中生成了100种不同丰度配置的序列读取数据。在我们的分析中,我们专注于具有有效多位点序列分型(MLST)方案的多个同种菌株的物种(方法------"CAMI2菌株疯狂基准")。这一纳入标准导致了对五个物种的菌株水平分析:肺炎链球菌(174个基因组)、大肠杆菌(97个基因组)、肺炎克雷伯菌(47个基因组)、金黄色葡萄球菌(21个基因组)和屎肠球菌(21个基因组)。
在RMSE-log方面,ChronoStrain−T在所有物种中均显著优于所有其他方法(扩展数据图2)。对于在原始CAMI2挑战中使用的L1范数误差,没有任何一种方法能够在所有五个物种中同时表现出优越性能;在半合成结果中,L1指标也呈现出类似的模式(扩展数据图3)。L1和RMSE-log之间明显的性能差异出现是因为L1在很大程度上忽略了来自众多低丰度基因组的误差贡献。对于这些基因组,ChronoStrain−T在所有被分析的物种中均一致优于其他方法。鉴于真实微生物群落的组成丰度跨越多个数量级,我们建议在评估复杂微生物群落的丰度估计时,不要仅仅依赖于L1。
UMB纵向样本的解释性提升
UMB项目对两个队列中的31名女性进行了为期一年的监测,这两个队列分别是"rUTI"(过去一年中多次泌尿道感染)和"健康"(近期无泌尿道感染史)。每位参与者每月提供一次粪便样本,所有参与者在第一个月都从直肠和尿液样本中培养出菌落。对于被诊断出泌尿道感染的参与者,如果可能的话,在诊断当天还会额外采集尿液样本并进行菌落培养。除此之外,还有参与者自我报告的最后一次已知抗生素使用日期和感染日期的元数据。除了原始样本外,我们还增加了一种新的数据模式。对于rUTI队列中通过原始StrainGST分析识别出爆发的样本子集,对在MacConkey琼脂(有利于包括大肠杆菌在内的革兰氏阴性菌)上培养的粪便样本进行了测序。
我们将ChronoStrain和StrainGST应用于UMB研究中的所有31个时间序列(附图G1-G31),UMB参与者18的模型输出如图3所示。注意,ChronoStrain的菌株簇以"CS"为前缀,而StrainGST的菌株簇以"SGE"为前缀。对于这两种方法,我们都使用"ST"前缀,用各自的多位点序列分型(MLST)标签对簇进行了注释。有关UMB分析流程的详细信息,请参阅方法部分的"UMB大肠杆菌分析"。

图3. ChronoStrain和StrainGST对rUTI阳性参与者UMB18的输出结果可视化
a, d, 使用两种不同的度量计算菌株的系统发育亚树:标记特异性k-mer比例距离(a)和全基因组k-mer Mash距离(d)。簇用前缀"CS"或"SGE"标记,以表示各自的聚类方法。多位点序列分型(MLST)标签(Achtman大肠杆菌方案)用"ST"前缀表示。b, e, CS(b)和SGE(e)聚类方法在时间序列中检测到的菌株的散点图。不同的标记表示样本类型(粪便、粪便的MacConkey培养、尿液)。实线垂直线表示泌尿道感染诊断日期。虚线垂直线在顶部用字母标记(例如,"N"表示呋喃妥因),表示自我报告的最后一次已知抗生素使用日期。c, f, CS(c)和SGE(f)在粪便中估计的"总体"相对丰度的图。使用n=5,000个后验样本,ChronoStrain的阴影区域是95%的可信区间;中心(实线)是中位数。
ChronoStrain的输出结果(图3b, c)表明,最初的感染很可能来自一个A生态型/ST69菌株(第0天CS1831旁边的黄色实心圆圈)。经过多轮抗生素治疗后,CS1831在尿液中已无法检测到,但在胃肠道(GIT)中仍然存在。事实上,在粪便中,多个时间点检测到两个ST69簇(CS1831,黄色实线;和CS1259,黄色虚线),其中优势簇与尿液样本中检测到的相同,也是17个时间点中最丰富的菌株。
另一个显著的菌株簇是B2生态型/ST95簇CS286(红色虚线),它对抗生素的反应不同;这个簇在两个富集的MacConkey培养样本中都有重现(图3b中的X;培养特异性丰度估计见扩展数据图4)。参与者在第55天之前报告的初始剂量的呋喃妥因和未知抗生素未能从GIT中清除这种菌株;其丰度在10-5以上或接近10-5。在第三和第四轮抗生素(β-内酰胺抑制剂)治疗期间,所有B2生态型菌株的丰度都大幅下降至10-6以下。然而,第五轮抗生素是在第300天左右重新使用呋喃妥因,ChronoStrain识别出CS286(旧菌株)和CS198(新菌株,红色实线)的快速增长,CS198是一种之前未检测到的新优势菌株。这些结果表明,与呋喃妥因相比,β-内酰胺更有效地清除了GIT中的B2分类单元。这与之前的文献一致,表明呋喃妥因在宿主体内的生物利用度更高,因此与β-内酰胺相比,在GIT中的积累较少。
解读StrainGST的输出结果(图3e, f)时,可以看到相邻时间点以互斥的方式调用了两个A生态型、ST69簇。这种时间序列/时间不一致性(随时间交替出现/缺失)在ChronoStrain进行的联合分析中是不存在的(补充文本F)。此外,粪便样本分析表明,ST95菌株存在于第187天之前但不包括第187天,然而在同一天的时间点上,MacConkey培养样本却表明并非如此。在ChronoStrain的分析中,相应的主要ST95簇在该特定日期仍然被检测到,这表明我们方法的联合分析得出了正确的检测结果。缺乏跨样本的一致性使得评估不同菌株对抗生素的敏感性或确定新菌株的爆发变得困难。此外,缺乏可信区间(或置信区间)也削弱了StrainGST的可解释性。
在刚刚介绍的UMB18的分析中,我们将不同的菌株簇定义为在数据库标记序列上核苷酸同一性低于0.998的簇。选择这个阈值是为了与StrainGST在其原始工作中用于定义簇的核苷酸同一性水平相一致。为了展示我们方法的实用性和可解释性,我们进行了与上述相同的分析,但采用了更精细的聚类(图4)。该分析的阈值设定为1−10-10核苷酸同一性,有效地捕捉了我们标记上的单核苷酸差异。
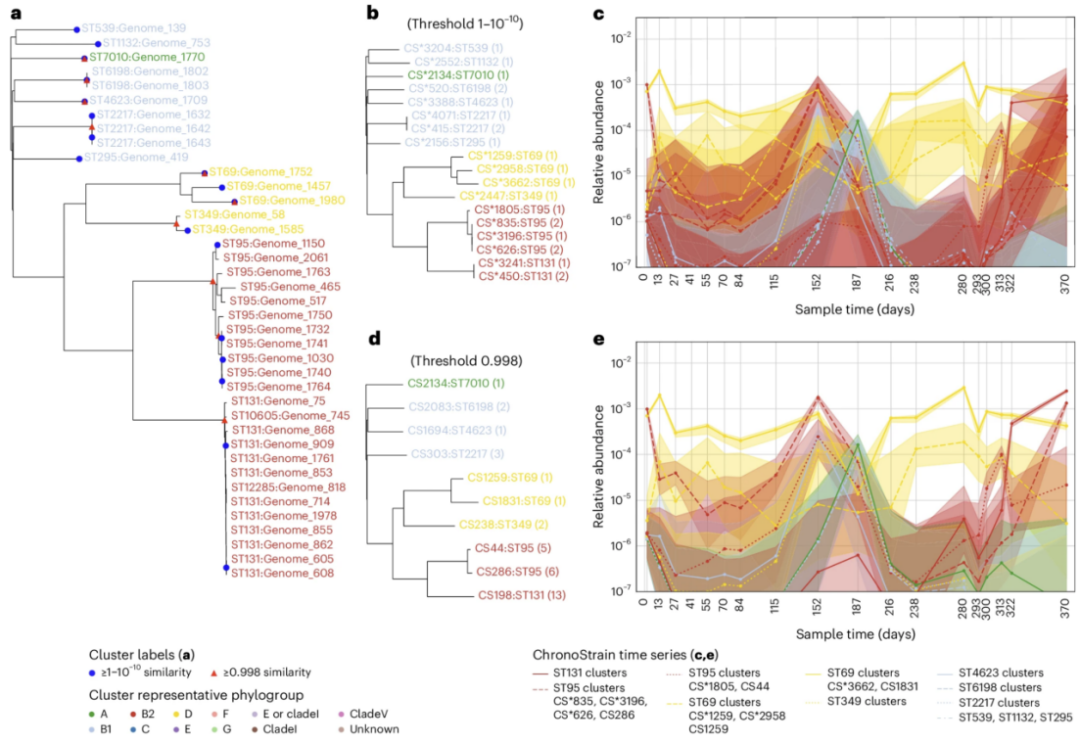
图4. ChronoStrain的可信区间是对不确定性的直接可视化
对UMB18的分析第二次运行时使用了一个更精细的数据库,聚类阈值设定为1−10-10相似性。这种精细的聚类用前缀"CS*"表示。a, 一个系统发育树,显示了UMB18在两种粒度下调用的簇。每个叶节点是一个基因组;内部节点表示每个簇的最近共同祖先。b, d, ChronoStrain在时间序列中调用的簇的阈值特定亚树,显示了精细聚类(b)和粗略聚类(d)。每个簇的大小在括号内:例如,"CS*835:ST95 (2)"表示簇CS*835包含两个基因组成分。c, e, 阈值特定的时间序列相对丰度,分别对应精细阈值(c)和粗略阈值(e)。多个轨迹的阴影区域(使用n=5,000个后验样本的95%可信区间)在时间上的近乎完美重叠表明需要对聚类进行粗化,例如在c中的ST95虚线红色中位数轨迹在e中合并在一起。中心线(实线)是中位数。实线和虚线黄色轨迹的不完美重叠表明存在两种不同的ST69菌株,其中一种的丰度比另一种高出一个数量级。
总体故事与之前相同:存在一个占优势的D类菌株,在时间序列的中间有一个B1类(或C类)菌株爆发,以及一个之前未检测到的B2类菌株在末尾成为优势菌株。然而,通过这种高分辨率的视图,我们确实调用了更多的菌株簇。然而,人们可以直接看到几种菌株的可信区间完全重叠,例如ST69簇的虚线黄色轨迹,或B1生态型簇的虚线蓝色轨迹。这表明模型在区分这些菌株方面存在困难,可能应该像在较粗的阈值中那样将它们聚类在一起。
婴儿样本的检测限提升
婴儿微生物组研究收集并测序了774名足月婴儿在新生儿期和婴儿期的纵向粪便样本,并从部分母亲那里额外收集了配对样本。在这项研究中,每个婴儿收集了1到6个粪便样本,大多数新生儿样本是在第4天、第7天和第21天采集的。从189名婴儿的粪便样本中获得了805个分离株。其中,349个分离株是粪肠球菌(321个来自婴儿,28个来自母亲)。我们将mGEMS流程以及ChronoStrain应用于这189名婴儿的时间序列粪便样本,并使用包含分离株基因组的数据库。
在确保两种方法的数据库处于同等水平后(方法------"BBS粪肠球菌分析"),我们对婴儿粪便宏基因组样本进行了推断。对于mGEMS,我们使用了参考文献中描述的相同超参数。为了在大致相等的灵敏度下比较这两种方法,我们调整了ChronoStrain的后验推断阈值,使两种方法报告的婴儿粪肠球菌分离株数量相同(图5)。
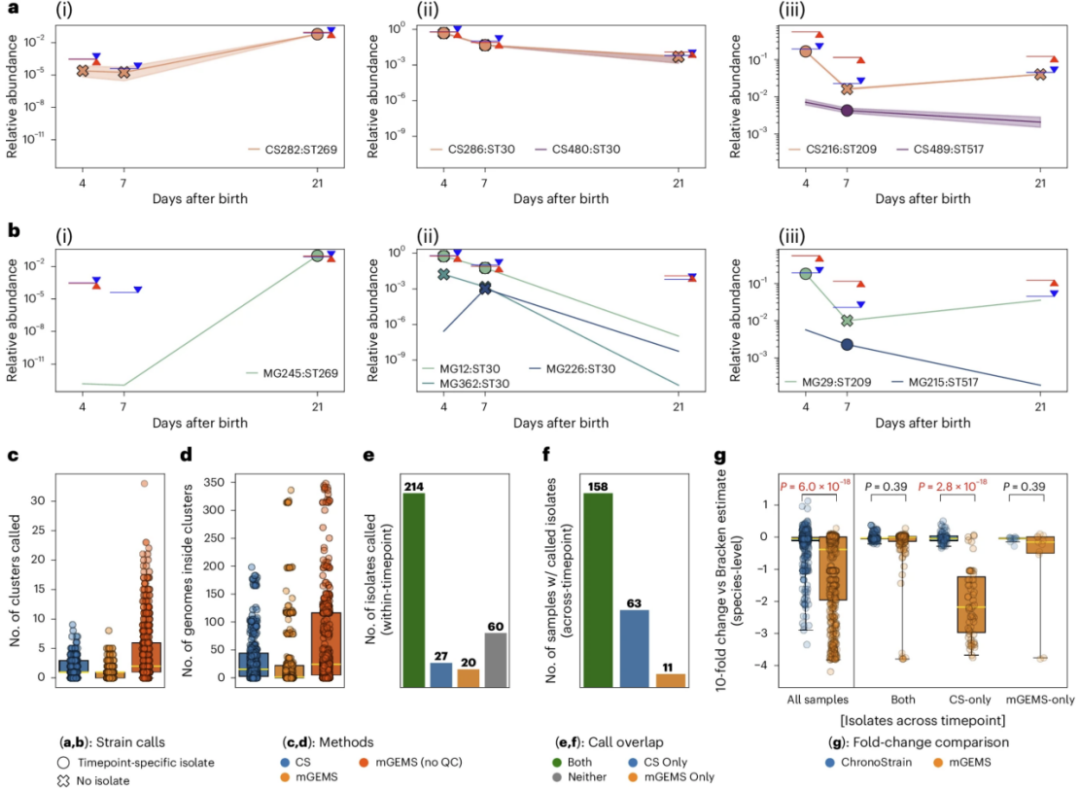
图5. ChronoStrain在时间上从婴儿样本中调用分离株,其丰度估计比mGEMS更准确
a, 婴儿A01077(i)、B00053(ii)和B02273(iii)的粪肠球菌菌株相对丰度估计示例。阴影区间是使用n=5,000个后验样本的95%可信区间。b(i--iii),相同婴儿的mGEMS估计值。对于每个簇,如果它在至少一个样本中通过了相应方法的过滤器,我们只绘制其轨迹。每个样本特定的菌株调用都根据该簇是否包含来自该供体样本的分离培养物而标记(实心圆圈/交叉)。蓝色三角形+水平线是Bracken粪肠球菌物种丰度估计值;红色三角形+线是MetaPhlAn4物种估计值。c, d, 在n=486个样本中通过过滤器的簇的数量(c)以及这些簇中包含的总基因组数量(d)。e, 从同一样本中调用的菌株数量(总共321个)。f, 通过任一方法或两种方法调用的分离株数量,其中分离株来自同一婴儿的"不同"样本,标记为"跨时间点"预测。g, 对于在f中分类的每个样本(例如,"两者"有n=158;"所有"有n=486),我们检查物种预测与Bracken的偏差程度。显示了经过Benjamini--Hochberg(BH)校正的配对双侧Wilcoxon检验的P值。在c、d和g中,中位数用黄色表示,箱体为25%和75%分位数,须线为2.5%和97.5%分位数。
正如预期的那样,每一样本的菌株调用数量(图5c)以及与同一时间点培养分离株相对应的菌株簇的数量(图5e)在两种方法之间相似。然而,我们确实注意到丰度估计存在显著差异。我们在图5a, b中用来自三个婴儿A01077、B00053和B02273的示例轨迹来说明这一点(完整集合见补充图H1-H21)。为了提供一个独立的比较,我们使用Kraken2+Bracken和MetaPhlAn4来估计粪肠球菌的物种丰度(图5a, b中的三角形)。mGEMS在BBS婴儿数据集中经常相对于Bracken产生低估(图5g,"所有样本"),在那些ChronoStrain对配对样本分离株进行菌株调用但mGEMS没有的样本中,Bracken和mGEMS之间的最大差异出现("仅CS")。为了更好地理解这种差异,我们绘制了每个样本相对于Bracken(和MetaPhlAn)的粪肠球菌丰度变化倍数(扩展数据图5)。在相对丰度约为0.01及以下时,mGEMS输出的粪肠球菌丰度为零(或接近零),与Bracken、MetaPhlAn和ChronoStrain不同。
最后,我们测试了当参考数据库不再包含与我们要追踪的菌株完全相同的基因组时,这些方法的稳健性。为了这个实验,我们突变了117个BBS分离株基因组,这些基因组已经被mGEMS调用(补充文本E.1),然后使用与之前相同超参数和阈值的两种方法进行推断。总体而言,mGEMS的调用从117/117减少到只有45/117个菌株,但ChronoStrain的结果从108/117基本保持不变,变为109/117(图6和扩展数据图6、7)。在补充文本E.2中,我们更详细地讨论了这些结果。
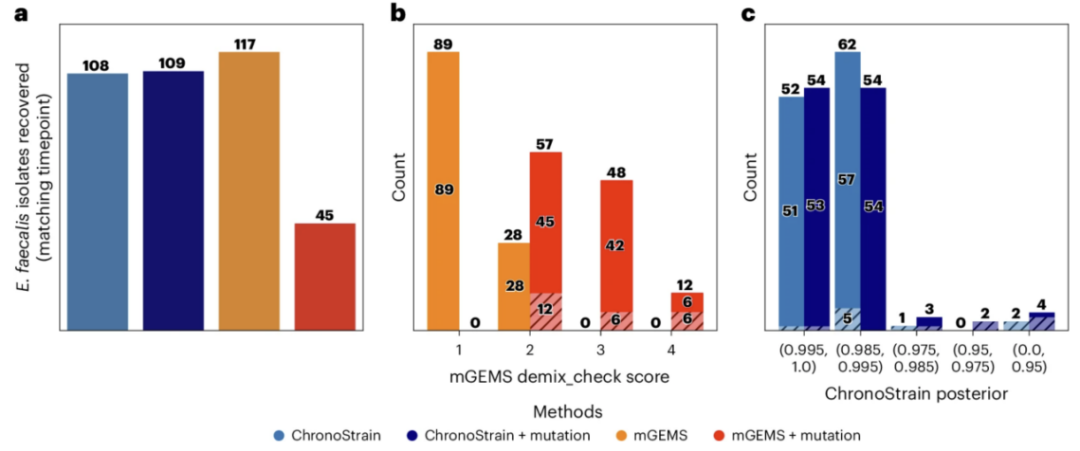
图6.与mGEMS相比,ChronoStrain对数据库基因组与样本序列读取之间的不匹配更具鲁棒性
我们在BBS数据上进行了推断,其中117个BBS分离株在被包含在数据库之前被突变(基因组突变率0.002)(补充文本E.1)。a, 每种方法调用的分离株簇的原始数量。注意,由于实验设计,mGEMS调用的分离株更多:这117个分离株最初是根据mGEMS的预测选择的,即使ChronoStrain没有调用它们。b, 所有117个分离株簇的demix_check分数分布;"1"为最佳,"4"为最差。每个条形分为两部分:实心上部区域表示丰度比≥0.01的菌株,对角线底部区域表示丰度比<0.01的菌株。突变的基因组导致demix_check分数急剧增加。c, ChronoStrain的后验概率;实心上部区域表示丰度比≥0.065的菌株调用,对角线底部区域表示丰度比<0.065的菌株调用。
我们强调,我们故意选择了那些已经通过mGEMS调用的配对样本的分离株,无论ChronoStrain使用未突变的数据库什么,这就是为什么我们报告了原始ChronoStrain运行的108/117调用,而不是完整的117个。mGEMS的demix_check诊断(该诊断试图衡量样本中基因组相对于数据库中基因组的新颖性)导致了菌株调用数量的减少:随着数据库中突变分离株的增加,分数变得更差(增加)(图6b)。可以通过允许更大的demix_check分数来增加正确的菌株调用数量,但这会以牺牲特异性为代价。当demix_check分数阈值从2增加到4时,mGEMS正确地调用了117个菌株中的93个,但每一样本的调用中位数比ChronoStrain多六倍(扩展数据图6c)。
这些实验表明,ChronoStrain对低丰度菌株的丰度估计更准确,并且在进行菌株调用时对数据库差异更具鲁棒性,而不会失去特异性。mGEMS对数据库中包含感兴趣菌株的分离和测序很敏感,且该流程不能可靠地估计低于10-2的丰度。这些差异可能会以统计学上显著的方式影响对菌株动态的解释。例如,在观察粪肠球菌的菌株更替(相邻时间点中最丰富的菌株不同)时,与mGEMS相比,ChronoStrain估计在第一个月内至少发生一次更替的婴儿数量是mGEMS的两倍(40/189对19/189,卡方检验P=0.0046,附表5)。
作者简介

Bonnie Berger (通讯作者)
● Bonnie Berger是麻省理工学院应用数学和计算机科学教授,也是麻省理工学院计算机科学和人工智能实验室计算和生物学小组的负责人。在麻省理工学院开始她的算法职业生涯后,她是计算分子生物学领域的先驱研究人员之一,并且与她指导过的许多学生一起,在定义该领域方面发挥了重要作用。Berger教授赢得了无数奖项,包括美国国家科学基金会职业奖和生物物理学会的戴霍夫研究奖。1999 年,Berger 教授被《技术评论》杂志评为首届 TR100 之一,成为 21 世纪顶尖的年轻创新者之一,2003 年,当选为计算机协会会员,并于 2010 年获得 RECOMB 时间测试奖。她最近当选为美国艺术与科学院院士,获得美国国立卫生研究院玛格丽特·皮特曼主任奖,当选为国际计算生物学学会 (ISCB) 院士,并获得洛桑联邦理工学院荣誉博士学位。她目前担任 ISCB 副主席、RECOMB 指导委员会主席以及 NIGMS 咨询委员会成员。此外,Berger教授还是布罗德研究所的准会员、HST的教员和哈佛医学院的附属学院。
信息来源:https://dms.hms.harvard.edu/people/bonnie-berger

Travis E. Gibson (通讯作者)
●Travis 在麻省理工学院获得控制理论博士学位,并在 NASA 和波音公司实习,然后将他的研究转向生物医学应用。目前在 BWH 和 HMS 担任主要教员,并在麻省理工学院计算机科学和人工智能实验室以及麻省理工学院和哈佛大学布罗德研究所担任其他职务。研究方向是利用机器学习和控制理论的工具来了解生物系统。
信息来源:https://comp-path.bwh.harvard.edu/travis-gibson/
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
iMeta高引 fastp PhyloSuite ImageGP2iNAP2 ggClusterNet2
iMeta工具 SangerBox2 美吉2024 OmicStudioWekemo OmicShare
iMeta综述 高脂饮食菌群 发酵中药 口腔菌群 微塑料 癌症 宿主代谢
10000+:扩增子EasyAmplicon 比较基因组JCVI 序列分析SeqKit2 维恩图EVenn
iMetaOmics高引 猪微生物组 16S扩增子综述 易扩增子(EasyAmplicon)
点击阅读原文