点击蓝字 关注我们
Cell Host & Microbe | Andrew J. Macpherson组-一项关于小鼠微生物群落中分类体-代谢关联的全球调查

研究论文
-
DOI:https://doi.org/10.1016/j.chom.2025.10.010
IF: 18.7 Q1 B1 -
原文链接:https://www.cell.com/cell-host-microbe/fulltext/S1931-3128(25)00424-X
-
第一作者:Bahtiyar Yilmaz
-
通讯作者:Andrew J. Macpherson(andrew.macpherson@unibe.ch)
-
主要单位:瑞士伯尔尼大学及其附属医院:伯尔尼大学医院内脏外科与医学系;生物医学研究系 Maurice Müller 实验室;瑞士联邦理工学院(ETH Zurich):分子系统生物学研究所;生物系统科学与工程系(巴塞尔);瑞士生物信息学研究所;苏黎世大学:进化生物学与环境研究系等
亮点Highlight
-
实验室与野生小鼠群体的肠道菌群组成存在显著差异
-
尽管分类学特征和栖息环境不同,其功能输出趋于一致
-
每个分类单元内存在多种变异,这些变异通常为特定饲养设施所独有
-
部分物种变异定位于核心代谢通路相关酶的活性位点
摘要Abstract
宿主与微生物群的互惠共生关系植根于饮食和代谢分子的交换。微生物多样性拓宽了代谢物库,不同类群以各异比例贡献着独特的化合物。在人体微生物组中,群落组成的高度可变性主要通过不同类群间相似的代谢功能得以补偿。然而,在多样性较低的小鼠模型中,这种补偿作用的程度如何,以及不同饲养设施是否具有代谢等效性,仍属未知。我们构建了一个可检索数据库,收录了全球51个小鼠饲养设施和12个野生小鼠种群的微生物组组成变异数据,其中饲养设施特有的变异已根据每种微生物物种的预测三维结构进行定位。我们配套的代谢组学数据表明,实际代谢潜力的变异性相对较低,为代谢补偿机制提供了功能性证据。此外,这种变异性与分类学组成而非饲养设施相关,揭示了可能与饲养设施间表型差异相关的类群-代谢物关联。整体而言,这一资源为加强微生物组研究和合作科学提供了重要工具。

结果Results
绘制实验室与野生群体中小鼠肠道微生物群的全球多样性图谱
作为探究实验小鼠饲养设施间微生物多样性深度及其功能影响的起点,我们通过分析来自48个不同机构的51个实验室群体的盲肠和粪便样本(覆盖全球范围),建立了一个反映小鼠微生物群组成异质性的资源库。鉴于扩展实验室小鼠可用模型性状和表型的重要性(这些小鼠已被自由生活的微生物群"野化"重定殖),我们还纳入了来自12个不同野生小鼠群体的样本(图 S1;表 S1)。正如预期,使用16S基因进行的属水平分析或通过宏基因组测序进行的物种水平分析均显示,每个饲养设施都具有独特的微生物群组成(图1和S2)。尽管不同文献报道的野生小鼠与饲养设施小鼠相比,其主要门类------芽孢杆菌门/拟杆菌门(原厚壁菌门/拟杆菌门 (F/B) 比率)的比例或高或低(这可能取决于所研究的不同野生小鼠群体以及实验室小鼠的不同供应商),但我们发现,在每个饲养设施内部以及采样的不同野生小鼠中,这些门类之间的比率范围都非常广泛(图1A)。饲养设施小鼠的微生物群通常与野生小鼠的微生物群组成不同,选定的饲养设施中存在的疣微菌门、支原体门(原柔壁菌门)和放线菌门,在所有研究的野生小鼠群体中仅以极低的比例存在(图1)。从这一广泛的地理数据集呈现出的图景,证实了全球范围内实验中使用的不同微生物群存在巨大的分类学异质性。该详细数据集可通过两个交互式Shiny应用程序访问,本文报告的所有分类学和代谢结果(https://yilmazlab.shinyapps.io/vivaria_app/)以及肠道微生物菌株的结构变异(https://yilmazlab.shinyapps.io/vivaria_app2/)均可根据饲养设施或野生群体进行搜索和探索。
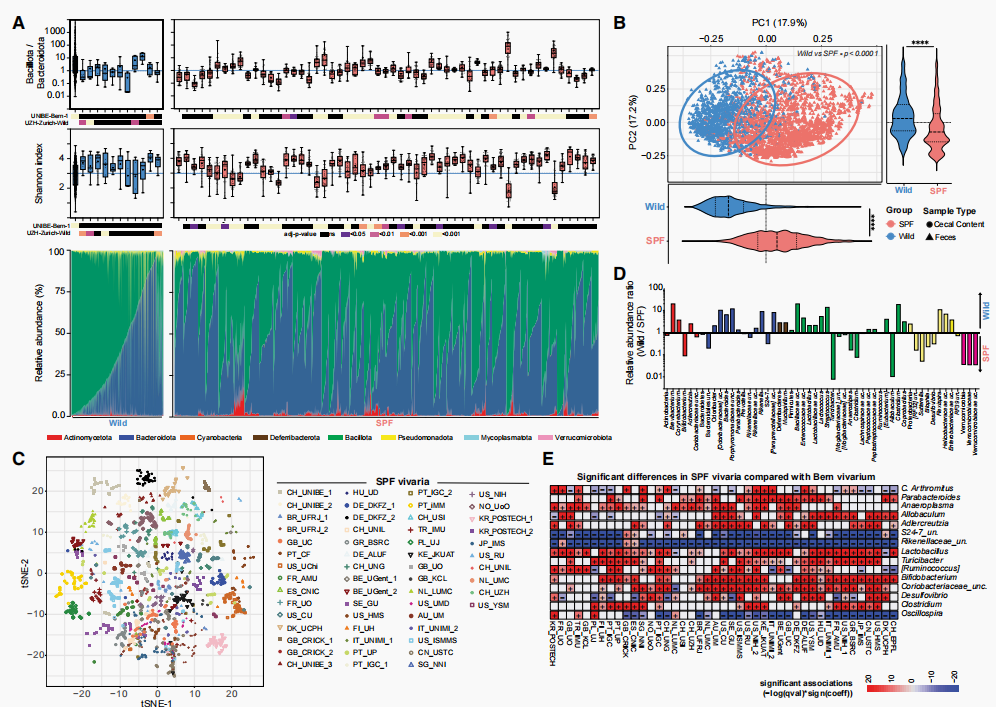
图1 | 全球范围内SPF与野生小鼠的肠道微生物群总体概况
(A) 基于16S rRNA基因测序数据集计算得出的芽孢杆菌门:拟杆菌门比率(上部)、香农指数(中部)和相对丰度(%)(下部),分别显示野生小鼠群体及不同SPF小鼠饲养设施的结果。每个箱线图代表一个采样点。每个饲养设施或野生小鼠群体均与CH_UNIBE或UZH_Zurich_Wild进行比较。热图显示调整后的p值。
(B) 基于Bray-Curtis距离的主坐标分析(PCoA)图,描绘了野生与SPF小鼠间的微生物群落差异。椭圆表示95%置信区间。通过Adonis检验对PCoA图进行组间差异分析(p < 0.0001),并对PC1和PC2的排名(±SD)进行Mann-Whitney U检验,其中****p < 0.0001。
(C) 使用t分布随机邻域嵌入(t-SNE)将所有来自不同饲养设施(来源)的微生物群样本投影至二维空间。
(D) 显示了所有野生小鼠群体与所有SPF小鼠饲养设施样本之间具有显著差异的细菌丰度(调整后p值 < 0.05)。
(E) 不同SPF饲养设施间微生物群组成的显著分类单元差异(调整后p值 < 0.05)。广义线性混合模型以伯尔尼大学群体(Bern 1)为参照运行。
(D和E)组间差异使用MaAsLin2确定。
另见 https://yilmazlab.shinyapps.io/vivaria_app/。
基因组多样性定义跨饲养设施和野生群体的小鼠肠道微生物群
尽管测序工作广泛开展,但人类和小鼠的测序分类单元中仍存在高比例的未定义分类单元及亚菌株变异。我们对全球范围内饲养设施和野生小鼠群体的广泛采样,使我们能够探究即使是从常规用于模拟健康和疾病的无特定病原体(SPF)饲养设施获取的序列信息也是多么不完整。我们优先选取了来自四个国家、16个饲养设施和12个不同地点的野生小鼠样本进行宏基因组测序(图2)。使用三种不同分箱工具(即 MetaBAT、19 MaxBin20 和 CONCOCT21)获得的宏基因组组装基因组(MAGs)被整合,以消除重复并提高基因组组装的准确性。根据MAG最低信息标准(MIMAG),该流程产生的总计20,345个MAGs达到了中等质量(完整性≥50%且污染率<10%)。随后使用 PhyloPhlAn 流程从这些MAGs重建了物种水平的系统发育和分类的物种水平基因组分箱(SGBs)。SGB基因组被划分为不同的亚组以展示其各自的完整性和污染百分比(图2A-2C):完整性在50%至90%之间的被指定为中等质量;90%-99.9%为高质量;100%为完成。污染水平低或高分别对应0.001%-5%或大于5%。近完整基因组(图2C)与中等质量基因组相比,显示出较低的菌株异质性(表S2)。总体而言,49%的分箱为高质量,其中282个完全闭合,其中98个无污染(图2C;表S2)。额外的基因组统计(包括重叠群数量和N50、平均mash距离)支持了近完整MAGs相较于中等质量MAGs持续保持高质量(表S2)。
与预期一致,与饲养设施或野生小鼠样本相比,人类样本中可分配到已知SGBs(kSGBs)的基因组比例相对更高,而小鼠样本中大部分被分配到先前未识别的SGBs(uSGBs)(图2D和2E)。在两个野生小鼠群体地点,超过90%的分箱分类单元未被分配(图2D),并且SPF饲养设施中kSGB和uSGB的组成存在明显异质性(图2E)。SPF小鼠中先前未识别的uSGBs主要属于拟杆菌门,而野生小鼠在变形菌门(原 Proteobacteria)和芽孢杆菌门内也有uSGBs(图2E)。尽管饲养设施和自然野生小鼠的微生物群组成有重叠,但本分析中的许多已知和先前未识别的SGBs,要么在不同野生小鼠群体之间共享,要么在不同SPF饲养设施小鼠群体之间共享,因此在无监督聚类中很大程度上区分了野生小鼠和SPF小鼠(图2E)。因此,即使在实验条件严格控制的小鼠设施中,已分配但先前未知的分类单元仍占很高比例,其中一些仅在单个饲养设施中发现。
由于分类学和注释不完整是人类微生物群中众所周知的问题,我们将小鼠数据与人类样本进行比较以评估潜在重叠(图S2A-S2C)。该分析表明,从人类样本获得的已知和先前未知的SGBs,与小鼠样本几乎完全不同(>99%)(图2E、2F和S2D;表S2),这反驳了洁净小鼠设施可能意外受到人类人员污染的观点。
鉴于小鼠野化重定殖迄今为止仅基于少数起始微生物群进行,通过使用不同且多样化的野生联合体,可能仍有相当大的潜力来拓宽模型性状和表型的范围(图S3A和S3B)。我们证实了野生小鼠微生物群存在季节性变化(图S3C-S3E)。这包括夏季芽孢杆菌门与拟杆菌门的比率显著降低。驱动夏与非夏月份之间这些周期性变化的大多数分类单元,集中在5个特定的细菌目内,包括芽孢杆菌门中乳杆菌目和梭菌目的大量减少,以及拟杆菌门中拟杆菌目和弯曲菌目的显著增殖。兼性厌氧的肠杆菌目在夏季也显示出显著扩张。这些发现表明,野生小鼠的肠道微生物群不仅高度异质(取决于采样群体的地理位置),而且还经历显著的季节性重塑,在更温暖、更潮湿的夏季,严格厌氧的芽孢杆菌门、耐氧的机会性变形菌门减少,而拟杆菌门增加(图S3E)。这种组成可塑性很可能由消费者资源(微生物:碳源)和季节性饮食差异所驱动。
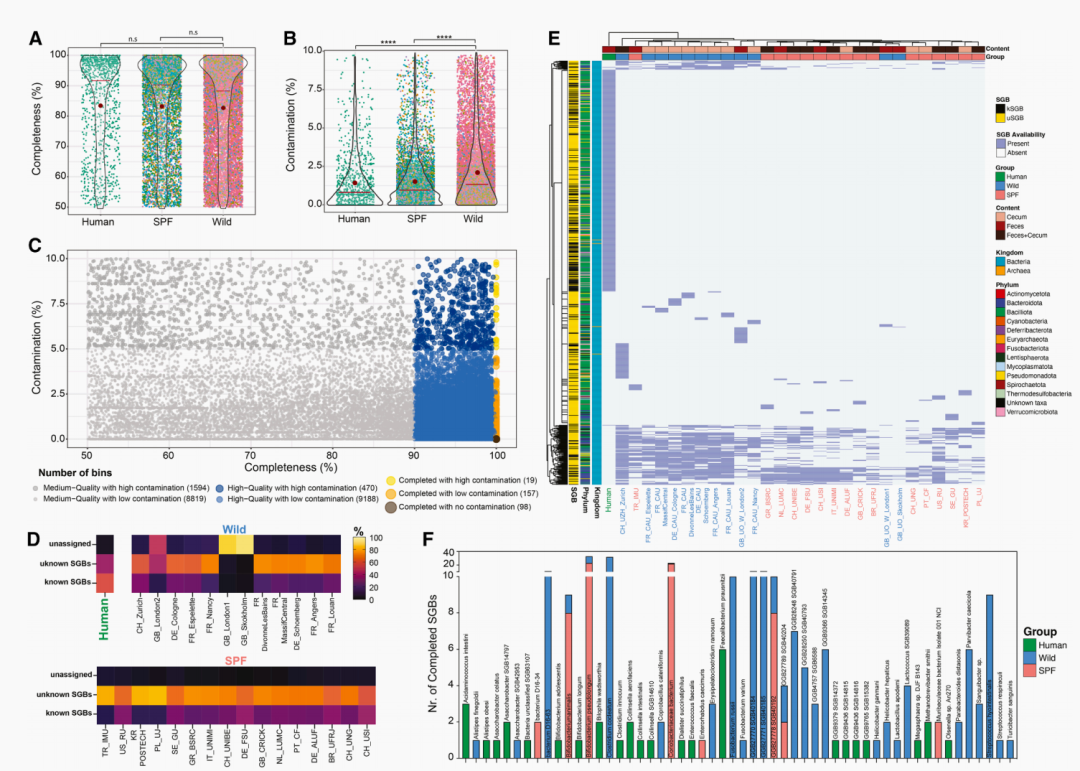
图2 来自全球实验小鼠饲养设施及野生群体的菌株宏基因组组装
(A和B) 宏基因组组装基因组(MAGs)生成自来自四个国家的16个SPF饲养设施和12个野生小鼠群体,其完整性>50%(A)且污染率<10%(B)。
(C) 显示了MAGs的分布情况,横轴为完整性,纵轴为污染率;每个点代表一个MAG,按群体着色(颜色随机,无特定含义)。
(D) 使用PhyloPhlAn流程获得的MAGs的物种水平基因组分箱(SGBs),显示了人类、不同野生小鼠群体(上部)以及不同SPF饲养设施(下部)中已知(kSGB)分类单元、未知(uSGB)分类单元和未分配SGBs的比例。
(E) 根据来源(人类、野生小鼠(苏黎世-NAA)或SPF小鼠(希腊-雅盖隆大学))分层,按kSGB或uSGB显示的亚菌株变异。
(F) 新完成且无污染的SGBs的来源与身份。条形图显示了样本来源:人类(绿色)、野生小鼠(蓝色)和SPF小鼠(红色),并显示了对应的身份信息。
人类与小鼠不同微生物群中的菌株水平遗传分化
微生物群中每个组成物种内部均存在遗传多态性。我们的目标是建立一个资源,用以阐明这些效应在不同微生物物种和小鼠群体间的表现,以及它们影响相关微生物联合体综合代谢产出的潜力。我们先前已在同生型小鼠中,通过遗传漂变和正选择证明了遗传进化现象,即个体细菌物种内部会随时间出现亚菌株。这些同生型小鼠是通过向C57BL/6无菌受体小鼠接种12个不同分类单元(每个起始于单个克隆)并在严格无菌条件下维持超过6年而创建的。以群体平均核苷酸一致性(popANI)> 99.9999%作为菌株同一性的指标,我们利用inStrain流程32对共存的基因组群体进行变异分析,旨在探究亚菌株在多大程度上导致每个群体内部或不同群体之间产生不同的多态性个体微生物物种,以及个体物种内的遗传变异在实验小鼠与野生小鼠之间如何比较。
我们从115只野生小鼠和115只实验室小鼠中分析了200种不同细菌物种的分类学重叠与遗传异质性。其中,48个分类单元在不同个体和/或小鼠间显示出超过95%的遗传相似性,这标志着物种边界,其覆盖度重叠范围在70%至99.9%之间。作为对照,我们在分析中纳入了健康人类样本,结果表明人类与小鼠之间共享的分类单元极少,或者这些分类单元是实验室小鼠或野生小鼠所独有的(图3A)。物种内高度一致的菌株(定义为99.9999% popANI,对于存在两个等位基因的情况,仅考虑主要变异)通常仅来源于单个饲养设施(图3B--3E)或单个野生小鼠群体内部(图3D和3E)。在比较实验室小鼠与野生小鼠之间,或人类与野生或实验室小鼠之间时,几乎未发现相同细菌分类单元内有完全相同的菌株。
有12个不同的分类单元在不同野生小鼠群体之间或不同SPF小鼠群体之间普遍共享(平均popANI范围在97%至99.5%之间),但强烈的遗传同一性(popANI > 99.999%)仅存在于每个独立的饲养设施或特定野生小鼠地点的分类单元内(图3E)。在饲养设施内部,拟杆菌门和芽孢杆菌门的物种以及肠杆菌科内广泛存在微多样性(popANI > 97%)。然而,仅存在少数例子显示物种内密切相关的菌株(ANI > 97%)出现在不同饲养设施之间(如 Akkermansia muciniphila、Turicimonas muris 和 Muribaculum sp002492595;图3C)或不同野生群体之间(如 Helicobacter cinaedi 和 Muribaculum sp001701195;图3C)。一个有代表性的系统发育树进一步突显了野生与SPF小鼠间共享的一个菌株的遗传相似性(图3D)。作为对比,在不同人类粪便样本中验证了不同宿主之间存在微多样性(97%--99.5%)35(图3F),尽管只有 B. uniformis 在人类与SPF小鼠样本间显示出遗传相似性,并且仅有六个人类菌株与野生小鼠群体的popANI重叠度 >97%(图3G)。这些结果表明,微生物群内个体分类单元的遗传同一性高度局限于每个独立的实验小鼠饲养设施或特定的野生小鼠群落。
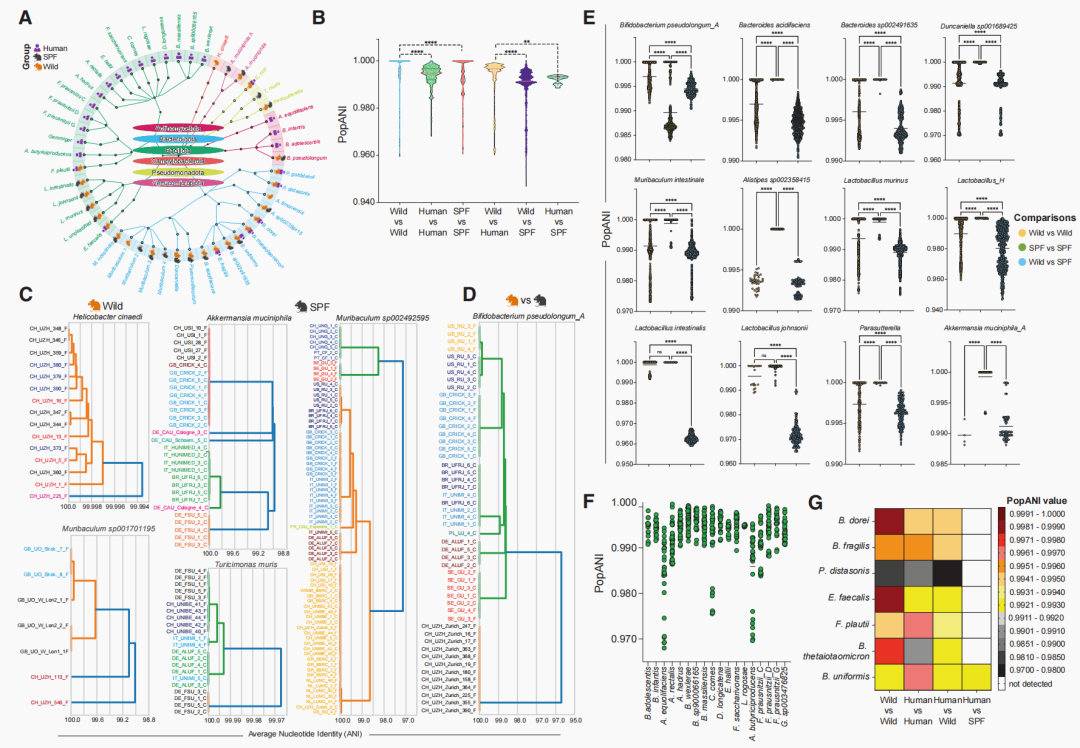
图3 | 小鼠与人类宿主肠道微生物群遗传同质性的比较分析
(A) 树状图显示了人类、野生小鼠与SPF小鼠间存在遗传相似性的微生物菌株。树状图中的颜色编码表示所识别分类单元的门级分类。
(B) 小提琴图展示了各组的总体群体平均核苷酸一致性(popANI)。数据报告涵盖种间和种内两个水平。
(C) 描绘了仅在野生小鼠群体或SPF饲养设施内观察到的遗传一致性微生物分类单元。
(D) 代表性系统发育树图,展示了某一特定菌株在野生与SPF小鼠间的遗传相似性。
(E) 以比较类型进行颜色编码的popANI值点图,显示了野生小鼠群体间、SPF小鼠群体间以及野生与SPF小鼠间的细菌物种比较分析。
(F) 点图说明了仅在人类中发现的菌株间的遗传差异。
(G) 热图显示了小鼠与人类样本间的菌株水平遗传相似性。显著性:* 调整后p值 < 0.05,** 调整后p值 < 0.01,*** 调整后p值 < 0.001,**** 调整后p值 < 0.0001;n.s.,不显著。
适应性代谢途径演化塑造多样化微生物群的功能分化
鉴于我们发现微生物菌株变异在很大程度上是特定于单个饲养设施的,并且我们之前已报道过在一个近交小鼠群体中微生物群分类单元随时间进化的证据,我们进而提出疑问:不同饲养设施中的个体分类单元是否独立于该分类单元在联合体中的丰度水平,已经发展出可能与酶或转运蛋白的代谢补偿相关的遗传变异性?由于非同义变异过多可以为该位点的正选择提供间接证据,我们推断,我们将能够检测到预测对微生物代谢关键步骤中酶的功能或蛋白质结构至关重要的非同义变异。
我们特别关注在饲养设施间表现出微多样性(以popANI值在97%至99%之间为特征)的微生物物种。在完成个体微生物物种内的基因注释后,我们进行了系统筛选,以识别在至少三个不同的饲养设施中表现出dN/dS比率大于一的变异。随后,我们选取了四个例子(图4A)结合AlphaFold3三维结构重建进行详细的结构分析(图4B-4J;https://yilmazlab.shinyapps.io/vivaria_app2/)。首先,通过检查TonB依赖性受体P3,我们观察到非同义变异在结构上受到限制,主要发生在预测的外部多糖利用结构域。同义变异也集中在该结构域,这可能是由于翻译约束所致,仅有一个变异位于形成蛋白质孔道的跨膜β-折叠片层中(图4I和4J)。其次,非同义变异也出现在高丝氨酸激酶中苏氨酸合成关键步骤的预测活性位点(图4D和4F),以及氨基磷酸核糖转移酶中从头嘌呤合成起始步骤的预测活性位点(图4G和4H)。非同义变异还聚集在高丝氨酸激酶(图4D和4E)和GTP环化水解酶1型(图4B和4C)四级蛋白质结构的预测亚基界面处。这些非同义变异中有许多是饲养设施特异性的,并且未出现在其他饲养设施的相同基因中,即使考虑到了所获序列深度的限制。虽然不同实验饲养设施之间共享的变异可能是由于动物及其微生物群在育种者和不同实验机构之间转移所致,但我们也发现了一些饲养设施小鼠和野生小鼠之间在非同义变异上的重叠,这支持了它们通过正选择独立衍生的观点(例如,氨基磷酸核糖转移酶------从头嘌呤合成的关键步骤;以及GTP环化水解酶------叶酸和生物蝶呤生物合成途径中的关键酶)(图4B和4C)。尽管这些变异尚未经过生化验证,但足够深度的正向遗传学方法可能有助于理解单个微生物群联合体内每个分类单元特异性代谢途径的功能。

图4 | 对饲养设施特异性微生物菌株变异的结构与进化解析揭示了选定关键功能基因中存在正选择证据
(A) 四个关键基因在野生和SPF小鼠不同饲养设施中dN/dS比率的分布。每个点代表一只个体小鼠,突出了饲养设施内部和之间的变异。这四个基因来自不同的微生物来源:A. muciniphila A的GTP环化水解酶(GTPCH)、B. pseudolongum的高丝氨酸激酶(HSK)、Bacteroides sp002491635的氨基磷酸核糖转移酶(ATase)以及B. acidifaciens的TonB依赖性受体P3。
(B) GTPCH催化反应的示意图,GTPCH是叶酸和生物蝶呤生物合成途径中的关键酶。
(C) 使用AlphaFold3生成的GTPCH环状结构。根据蛋白质-配体相互作用分析工具(PLIP)预测可能参与多聚体化的非同义替换位点以红色高亮显示。呈现这些替换的饲养设施样本在附加热图中以深蓝色标出。红色字体的饲养设施代表SPF小鼠,蓝色字体的代表野生小鼠。
(D) HSK催化反应的示意图,HSK是苏氨酸生物合成途径中的限速酶。
代谢灵活性及资源驱动的特化区分野生与SPF小鼠的微生物群
考虑到个体动物间微生物组物种组成和基因组变异的巨大多样性,我们接下来评估了微生物群内不同的代谢功能是否理论上以不同的组合方式结合以实现代谢稳态。为了检验不同微生物群落的代谢潜力,我们首先使用HUMAnN3流程分析了来自不同饲养设施和野生小鼠群体的盲肠及粪便宏基因组鸟枪法测序数据,以探究微生物代谢途径。结果显示,与SPF小鼠相比,野生小鼠的粪便和盲肠样本中途径丰富度均有所增加;与小鼠相比,对照人类样本的途径丰富度也更高,尽管存在重叠范围(图5A)。基于Bray-Curtis距离的主坐标分析(PCoA)证实,SPF小鼠中存在的83条注释代谢途径与野生小鼠的途径重叠,并且人类粪便样本中注释的途径聚集在靠近野生小鼠盲肠和粪便样本所呈现的广泛β-多样性分布区域(图5B)。这些结果证实,尽管微生物群存在巨大差异,但其累积代谢潜力存在重叠。
使用MaAsLin2进行的统计分析显示,野生小鼠、SPF小鼠和人类之间具有广泛共享的途径代表性,没有更高层次的聚类能独特地区分任何一组(图5C)。来自18个SPF饲养设施中6个的盲肠样本聚集在一个主要与对照健康人类粪便样本共享的组中,尽管人类微生物群落特别包含糖原降解途径(图5C宿主聚类2,途径PA233)。主要包含野生小鼠的聚类(图5C,宿主聚类1和3)的特征是,注释为从头核苷合成途径及其他寡糖合成的基因代表性增加,这可能是因为与人类和饲养设施小鼠的自由采食相比,野生小鼠的碳源限制更大(图5C;表S3)。微生物群对从头核苷合成的需求取决于用于源自细胞更新或饮食来源的能量高效补救途径的核碱基生物利用度。尽管我们无法评估所采样野生小鼠的饮食或食物偏好,但植物材料是核苷酸的贫乏膳食来源,并且我们确认了饲养设施饲料中存在膳食核苷酸(图S4;表S4)。
因此,尽管每个饲养设施(图1C--1E)和野生群体(图S3B)的微生物组分类学组成不同,但对注释基因内容的代谢潜力变异性的无监督分析并未根据饲养设施或野生与实验室小鼠进行聚类(图5C)。推断的累积代谢潜力的大多数特征甚至未能在不同宿主组间分离。这反映了跨环境的微生物物种之间存在显著的功能补偿,使得多样化的分类学消费者组合能够在特定饮食条件下利用资源可用性。
尽管我们优先选择了来自18个饲养设施的肠道样本进行宏基因组测序,但我们还拥有来自51个饲养设施、12个野生小鼠群体和107个人类样本的16S基因测序数据(另见https://yilmazlab.shinyapps.io/vivaria_app)。因此,我们测试了是否能在更广泛的样本收集中找到这三个群体具有不同代谢潜力的证据。由于缺少模型拟合所需的注释,一些样本被排除在此分析之外(见STAR方法)。分析的样本覆盖了21个饲养设施、1个野生小鼠群体和105个人类样本。我们通过先前发表的一组基因组尺度代谢模型(GSMMs)45 将扩增子序列变异(ASVs)映射到代谢反应,并利用此映射量化每个样本中的反应丰度(图S5E--S5I)。该映射产生了7,604个独特的代谢反应,其中3,146个可以映射到京都基因与基因组百科全书(KEGG)中的134个不同代谢子系统之一,以及一个用于无法映射到已识别子系统的反应的"未知"子系统。然后,通过拟合反应特异性线性混合效应模型,将所有饲养设施(图S5E--S5I)、野生群体和人类样本之间的反应丰度差异与感兴趣的组相关联。我们估计了感兴趣的组之间反应丰度的平均差异,并使用Holm-Bonferroni多重检验校正测试了显著性(调整后p值 ≤ 0.05)。对于每个感兴趣的组,然后使用Fisher精确检验(p ≤ 0.05)检验了每个代谢子系统中显著反应的富集情况。映射到未知子系统的反应在富集检验中被考虑,但未知子系统本身未被检验。正如预期,将仍在哺乳期(摄入含乳糖和牛奶糖蛋白的乳汁)的幼鼠与成年小鼠进行比较时存在明显差异。这些差异在微生物水平(图S5A--S5D)和代谢水平(图5D)均可见。诸如半乳糖代谢、糖酵解、肽聚糖生物合成和甘油磷脂代谢等子系统在幼鼠中平均丰度显著高于成年小鼠的反应中富集(图5D)。然而,与对不同微生物群累积代谢潜力的宏基因组分析一致,野生和SPF小鼠的代谢存在显著差异,特别是在野生小鼠中更高的色氨酸代谢、吲哚生物碱生物合成和芥子油苷生物合成方面(图S5G)。与小鼠相比,对照人类中少数代谢途径的平均反应丰度相对更高,包括参与缬氨酸、亮氨酸和异亮氨酸生物合成、赖氨酸生物合成、脂肪酸生物合成和延伸的途径(图S5E--S5I)。尽管不同小鼠微生物群之间存在普遍的代谢补偿,但子系统代表性的差异出现在不同生命阶段(可能是由于哺乳期与断奶后饮食的消费者-资源效应所致),并且可能在野生与饲养设施小鼠之间也存在。
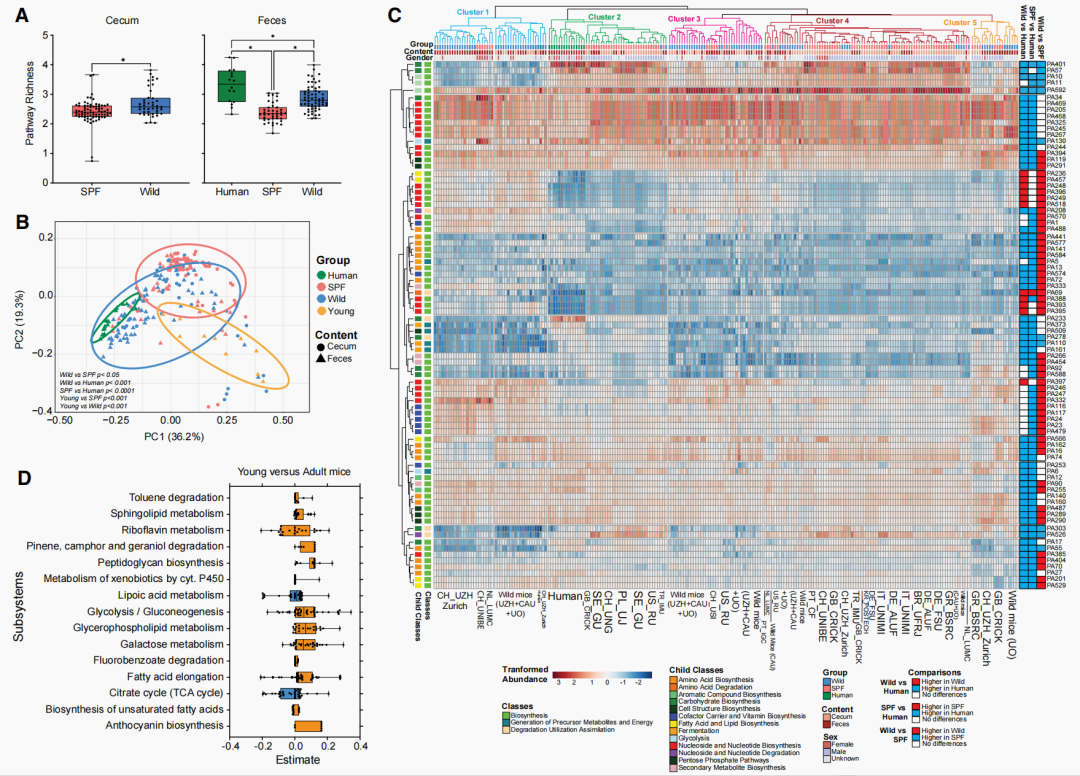
图5 | 小鼠与人类微生物群落的代谢潜力
(A) 箱须图显示了使用HUMAnN3流程分析得出的野生小鼠、SPF小鼠与人类样本间途径丰富度的差异(展示四分位数、范围及标准差)。*p < 0.05。
(B) 基于Bray-Curtis距离的主坐标分析(PCoA)图,展示了野生小鼠、SPF小鼠与人类样本间推断的微生物代谢途径。各组的位置由椭圆表示(95%置信区间)。分别采用非参数Mann-Whitney U检验和Adonis检验分析α多样性(A)与β多样性(B)的组间统计差异。
(C) 使用MaAsLin2识别了与不同组别存在显著关联、且在至少30%样本中未检出的微生物代谢途径的相对丰度。热图显示了样本类型间相对丰度存在显著差异的推断微生物代谢途径(调整后p值 < 0.05)。途径相对丰度通过颜色标度表示,从低(蓝色)到高(红色)。
(D) 基于基因组尺度代谢模型/混合效应模型分析,估计了由小鼠年龄差异(成年/幼年)引起的、在显著富集的子系统(p ≤ 0.05)中平均反应丰度的显著差异。每个点代表一个估计值,仅显示显著估计值(调整后p值 ≤ 0.05)。正值表示该子系统在幼鼠中富集。
另见 https://yilmazlab.shinyapps.io/vivaria_app/。
补偿稳定性与代谢物敏感性揭示多样化微生物群的功能适应性
通常,通过宏基因组分析或ASV的代谢反应映射来解读微生物群的功能性代谢补偿存在局限性,因为这些方法仅提供代谢潜力的估计,而根据实际通量的不同,可能导致不同的代谢组结果。与其依赖对代谢潜力的间接估计,实验者所需的关键信息在于了解已实现的代谢物组成,即微生物联合体组成的变化可能如何导致其腔内浓度的显著偏离。为了评估这一已实现的组成,我们测定了不同饲养设施(62只成年SPF小鼠,10-18周龄)、野生小鼠(9只)和无菌小鼠(5只)盲肠内容物中的稳态代谢组组成。对112种典型肠道代谢物的靶向分析显示,在不同饲养设施的动物盲肠内容物中,除无菌小鼠(正如预期,其代谢物多样性降低)外,所有广泛的化合物类别均呈现相似的分布情况(图6A, 6B, S6A, S6B;表S5)。对此数据的排序分析显示主成分方差较低(≤12%),仅无菌小鼠被显著区分开来(图6C)。在无监督分析中,小鼠也未按饲养设施分离(图6D;表S5)。
为了评估微生物组组成以及饮食如何与已实现的代谢物组成相关联,我们重点关注了47只具有匹配的宏基因组学、腔内代谢组学和(饲养设施特异的)饮食代谢组学数据的小鼠(42只SPF,5只野生,来自10个饲养设施)。我们首先单独分析了三种数据类型的变异性(图S6C)。跨小鼠的腔内代谢物浓度变异性,以单个代谢物的变异系数(CV)表征,其中位CV为179%(四分位距IQR 138%, 258%),远高于典型的技术变异性(约10%)。由于跨饲养设施的饮食代谢物变异性处于同一量级(中位CV:130%,IQR 96%, 186%),我们检验了跨饲养设施的腔内与饮食代谢物丰度变异性之间是否存在显著差异(单侧F检验,α = 0.05)。图6E的结果并不支持腔内代谢物存在系统性的更高变异性,这表明在已实现的代谢组成中也存在功能补偿。
随后,我们使用线性混合效应模型来量化分类学(71个在物种水平鉴定的分类单元)、饮食(124种代谢物)和代谢组组成之间的关系。具体来说,我们选择了最能解释腔内代谢物变异性(就饮食和物种组成而言)的每代谢物模型(详见STAR方法)。仅包含饮食预测因子的模型所解释的变异性显著少于包含物种预测因子的模型,且它们未能产生最优的每代谢物模型,后者均显示出较高的解释力(R² ≥ 0.9;图S6D)。少数显著预测因子与饮食相关,且这些因子的估计效应较小(图S6E)。饲养设施或微生物群模型未被识别为显著因子(表S6),这证实了物种组成主要塑造腔内代谢组。图6F展示了跨代谢物的估计且显著的消费者和生产者关系;此处,颜色表示估计的代谢物生产或消耗,颜色强度反映了物种排除时模型R²的变化。许多模型包含了多个物种(生产者和消费者群体)的贡献。这表明,至少对于这些代谢物而言,肠道代谢环境可能是广泛的交叉取食与协作的结果,而非由少数特化的"关键"物种决定。物种丰度与对腔内代谢组的估计效应数量或强度之间缺乏相关性(图S6E),支持了这一观点。最后,对于许多模型,分析突出了一组具有高估计效应的推定贡献物种;这可能是由于实际的代谢特化所致,也可能与在物种水平鉴定的分类单元份额有限有关,或与其他因素(如未建模的物种间相互作用)有关。我们得出结论:已实现的代谢潜力其变异性与功能补偿相符,并且这主要可以通过分类学组成以及分类单元间广泛的代谢相互作用来解释。
为了进行更深入的分析,我们聚焦于琥珀酸,它被认为是微生物群复杂碳水化合物发酵的产物,并且是下游短链脂肪酸(SCFA)生产的碳源。鉴于SCFA和琥珀酸在宿主生理和免疫中的普遍重要性,我们分析了这些分类单元中琥珀酸生产和消耗的注释途径,以及基于GSMM预测的它们生产和消耗琥珀酸的能力(见STAR方法)。这两种对代谢潜力的表征与线性混合效应模型推断的贡献高度一致(图6G),表明来自匹配代谢组学和宏基因组学数据的推断能够为微生物组中的生产者-消费者关系提供有效的假设。
代谢物浓度反映了由饮食、宿主代谢物脱落和微生物代谢共同塑造的稳态。尽管饲养设施与野生群体之间存在明显的分类学变异,代谢组谱与宏基因组学及基因组尺度建模结果相符,表明累积的代谢潜力在很大程度上补偿了个体差异。然而,补偿的程度取决于化合物、类别、饮食和环境背景。值得注意的是,不同分类单元对功能性代谢结果的贡献存在差异,这无法仅从其编码的潜力推断出来。

图6 | 无菌、野生及SPF小鼠的肠道代谢谱
(A) 基于MetaCyc注释,对来自无菌小鼠、野生小鼠及13个SPF饲养设施的62份盲肠样本中丰富代谢物进行的分类。条形图显示了各类别的平均相对丰度。
(B) 无菌、野生及SPF小鼠代谢物丰富度的箱线图(展示四分位数、范围及标准差)。**p < 0.01。
(C) 基于Bray-Curtis距离的主坐标分析(PCoA)图,展示了无菌、野生及SPF小鼠间的代谢物差异。
作者简介
Bahtiyar Yilmaz 第一作者
(第一作者)

Bahtiyar Yilmaz,瑞士伯尔尼大学内脏外科与医学系研究员,微生物组与宿主代谢互作领域专家。主导全球小鼠微生物组资源库构建与多组学整合分析,在菌株级进化和代谢补偿机制研究方面取得突破性成果。主持瑞士国家科学基金会(SNF)启动基金,参与多项欧盟研究委员会(ERC)项目。现任《Cell Host & Microbe》特约审稿人,致力于推动微生物组研究标准化与可重复性。
Andrew J. Macpherson 通讯作者
(通讯作者)

Andrew J. Macpherson,瑞士伯尔尼大学内脏外科与医学系教授,微生物组与免疫发育研究权威。长期致力于宿主-微生物互惠共生机制解析,在无菌动物模型和微生物组移植技术领域贡献卓著。主持瑞士国家科学基金会Sinergia项目及欧盟研究委员会HHMM-Neonates项目,获多项国际微生物组研究奖。现任《Nature Microbiology》编委,主导跨国微生物组资源整合计划。
翻译:刘家汇,西北农林科技大学
审核:朱志豪,广东医科大学,基因组所联合博士后
终审:刘永鑫,中国农科院基因组所,研究员/博导
排版:申子昂,华南理工大学,基因组所联培硕士在读
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
iMeta高引 fastp PhyloSuite ImageGP2iNAP2 ggClusterNet2
iMeta工具 SangerBox2 美吉2024 OmicStudioWekemo OmicShare
iMeta综述 高脂饮食菌群 发酵中药 口腔菌群 微塑料 癌症 宿主代谢
10000+:扩增子EasyAmplicon 比较基因组JCVI 序列分析SeqKit2 维恩图EVenn
iMetaOmics高引 猪微生物组 16S扩增子综述 易扩增子(EasyAmplicon)
点击阅读原文