论文题目:Deep contrastive learning enables genome-wide virtual screening
深度对比学习支持全基因组虚拟筛选(Yinjun Jia†, Bowen Gao†, Jiaxin Tan†, Jiqing Zheng†, Xin Hong†, Wenyu Zhu, Haichuan Tan, Yuan Xiao, Liping Tan, Hongyi Cai, Yanwen Huang, Zhiheng Deng, Xiangwei Wu, Yue Jin, Yafei Yuan, Jiekang Tian, Wei He, Weiying Ma, Yaqin Zhang, Lei Liu*, Chuangye Yan*, Wei Zhang*, Yanyan Lan*)DRUG DISCOVERY
个人总结:
核心是解决 "全基因组规模虚拟筛选太慢" 的痛点 ------ 提出基于深度对比学习的框架,将蛋白质口袋和小分子编码到同一特征空间,实现 "搜索引擎式" 快速匹配。
数据准备:从ProFSA中的伪配对+一点真实的配对
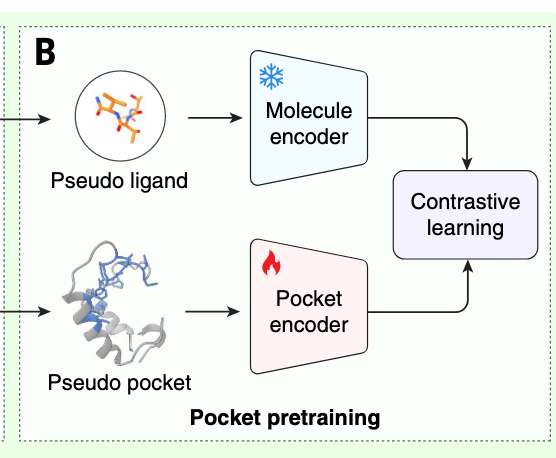
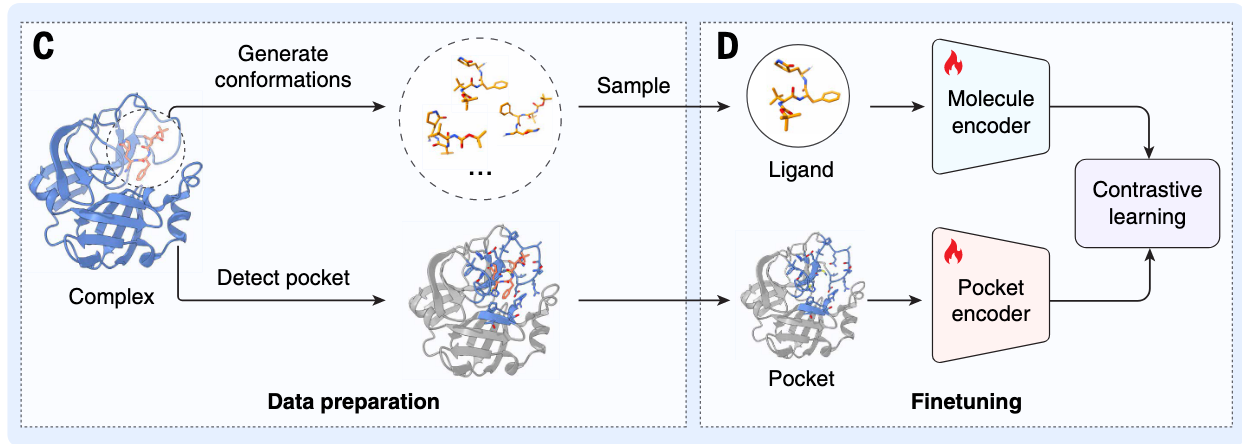
模型训练:预训练选择冻结分子编码器,训练口袋编码器(这里用到对比学习),微调时同时更新分子和口袋编码器对比损失。
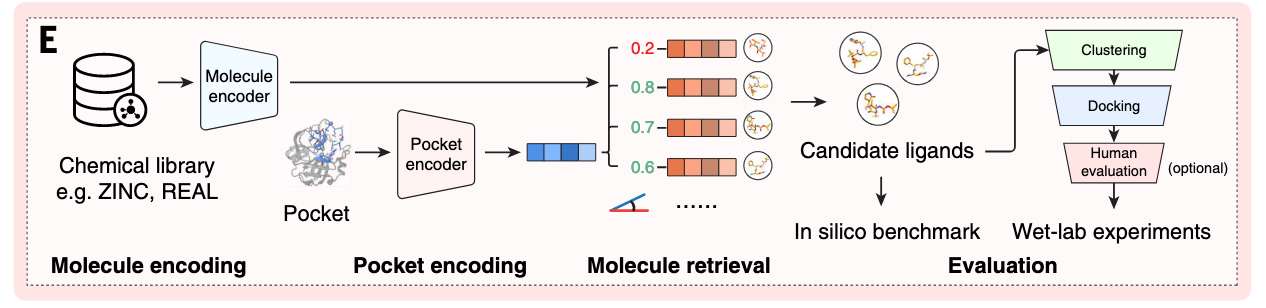
虚拟筛选:首先将口袋和化合物转成特征向量,去计算余弦相似度,最后用湿实验验证。
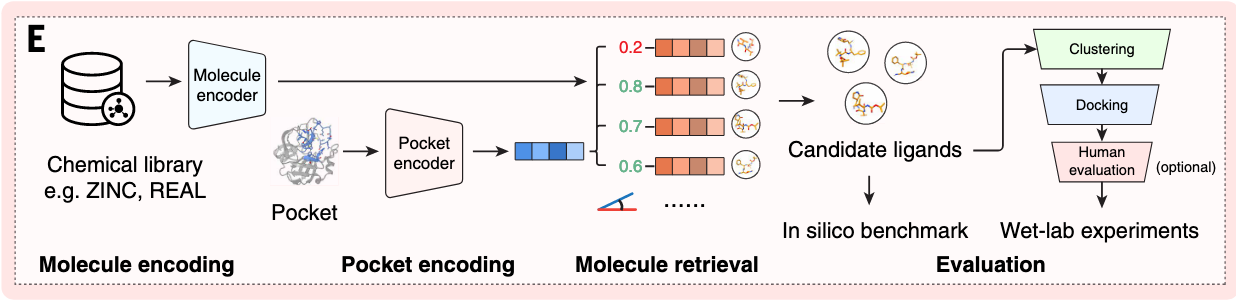
主要点:他把筛选变成检索大大加快速度,先把所有化合物提前编码,筛选的时候只需要目标口袋的信息,再用相似度排序。
与之前ProFSA论文相似点:都解决 "蛋白 - 小分子结合数据稀缺" 的问题(ProFSA 缺口袋表征数据,DrugCLIP 缺虚拟筛选数据)
不同点:一个是应用导向,一个是基础导向。
摘要
- 蛋白质结构预测的最新突破为全基因组药物发现开辟了新途径,但现有的虚拟筛选方法在计算上仍然存在限制。我们介绍DrugCLIP,一个对比学习框架,实现了超快且精准的虚拟筛查,速度高达对接的1000万倍,同时在计算机基准测试中持续优于各种基线。在湿实验室验证中,DrugCLIP实现了去甲肾上腺素转运蛋白15%的命中率,并确定了两种已鉴定的抑制剂结构与目标蛋白复合物。对于甲状腺激素受体相互作用体12号,该靶点缺乏全息结构和小分子结合剂,DrugCLIP仅使用alphaFold2预测结构,成功率达到了17.5%。最后,我们发布了GenomeScreenDB,这是一个开放访问数据库,提供对~1万种人类蛋白质的预计算结果,筛选对象为5亿种化合物,开创了后alphafold时代的药物发现范式。
引言
人类基因组由约20,000个蛋白质编码基因组成,其中许多与多种疾病相关。尽管如此,只有约10%的这些基因被美国食品药品监督管理局(FDA)批准的药物成功靶向,或文献中已记录小分子结合剂。这使得潜在药物基因组中相当一部分尚未被充分探索,这为治疗创新带来了一个充满希望的机会。科学界渴望将生物学相关靶点转化为药物突破。然而,大多数研究人员缺乏先进的高通量筛查设备或足够的计算能力来进行全面的虚拟筛查。此外,蛋白质通常作为家族或通路的一部分,表明单一蛋白质靶向可能并非总是最有效的策略。这些限制会显著降低药物发现的成功率,尤其是针对新靶点。因此,开发一个包含全基因组虚拟筛选结果的综合化学数据库,将是生物医学研究界宝贵的资产,有望显著加速新药的发现。
鉴于对所有人类蛋白质进行实验筛查所需的大量时间和资金成本,虚拟筛查已成为应对大量潜在靶点的更实用方法。 在经典的计算机辅助药物发现(CADD)中,分子对接是基于靶点的虚拟筛选的基础技术。尽管简化评分函数、优化算法和硬件加速有所进步,分子对接仍耗时,评估每对蛋白质-配体通常需要数秒到几分钟。例如,最近一次大规模对接行动需要2周时间,即使使用了1万个中央处理器核心,也需要2周时间将10亿个分子与单一靶标进行筛选。因此,全基因组虚拟筛查的计算需求极高,使得现有技术难以实现此类工作。
人工智能(AI)在药物发现方面具有巨大潜力。多种深度学习方法已被开发用于虚拟筛查,重点是预测配体-受体亲和力。然而,将这些方法应用于大规模虚拟筛查仍面临重大挑战。主要问题是由于实验条件异构导致亲和力值不一致,这可能对训练模型的性能产生负面影响。此外,训练数据集与现实测试场景之间的分布显著变化,阻碍了AI模型的普遍化,因为现实虚拟筛查通常涉及的非活性分子比例高于精心筛选的训练集。此外,深度学习模型拥有数百万参数的计算需求,尤其在化学库和目标数量增加时,成为推断速度的关键瓶颈。因此,迫切需要开发更高效、更稳健的人工智能方法,以有效应对这些挑战。
本研究介绍了DrugCLIP,一种虚拟筛选的对比学习方法 ,通过**比对蛋白口袋和配体表示来区分强效结合剂和非结合剂。**对比学习在多种应用中取得了成功,DrugCLIP将这些优势扩展到了药物发现领域。DrugCLIP不依赖结合口袋的准确局部几何,仅允许基于apo或计算预测结构进行结合预测。我们的模型在DUD-E(18)和LIT-PCBA(19)基准测试中取得了竞争性能,这两种广泛使用的虚拟筛选数据集,优于许多传统基于对接的方法和神经网络。它还通过鉴定经实验验证的5-羟色胺(血清素)受体2A(5HT2AR)、去甲肾上腺素转运蛋白(NET)和甲状腺激素受体相互作用因子12(TRIP12)的结合剂,展示了实际应用价值,后者缺乏已知的抑制剂和实验性全息结构。最后,我们利用DrugCLIP对AlphaFold2预测的人类蛋白进行了全基因组虚拟筛查(20, 21),结合位点由口袋检测算法定义(22, 23)和生成式人工智能模型,筛查了超过5亿种公共化合物(24, 25)。该虚拟筛选活动在24小时内使用八个图形处理单元(GPU)完成了超过10万亿次蛋白质-配体评分操作。最终生成的GenomeScreenDB包含了超过200万个潜在命中,分布在2万多个口袋中,涵盖约1万种蛋白质,覆盖了近一半的人类基因组。所有分子、分数和姿态均可在 https://drugclip.com 免费获取,便于进一步研究。
DrugCLIP模型的设计
与以往依赖回归直接预测蛋白质-配体亲和力值的机器学习模型不同,DrugCLIP(图1)将虚拟筛选重新定义为一项密集的检索任务。其关键创新在于其训练目标,即学习由独立神经网络编码的蛋白质口袋和分子的对齐嵌入空间。然后可以用向量相似度指标反映它们的结合概率 。在训练过程中利用对比损失,蛋白质口袋与其结合蛋白(正性蛋白-配体对)之间的相似性被最大化 ,而蛋白质口袋与分子结合其他靶点(负性蛋白-配体对)之间的相似性则被最小化。
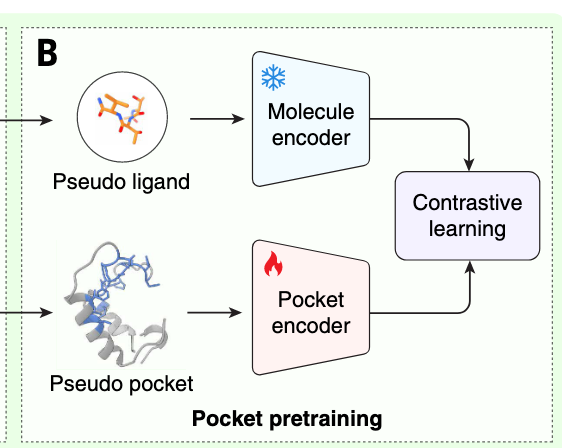
DrugCLIP的培训流程包括两个阶段:预培训和微调。分子和口袋编码器会先用大规模合成数据预训练,并在微调过程中通过实验确定的蛋白质-配体复合结构进一步完善。在预训练阶段,分子编码器初始化为Uni-Mol,这是一种成熟的分子编码器。分子编码器冻结后,口袋编码器被随机初始化并训练,通过对比学习与分子编码器对齐(见图1B)。
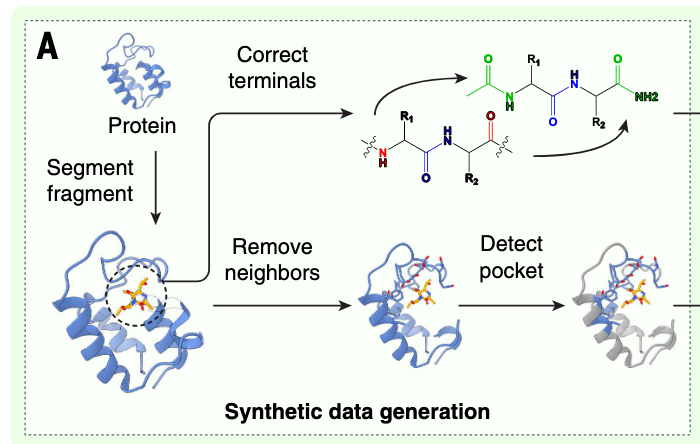
我们开发了一个蛋白质片段-包围比对(ProFSA)框架(见图1A),用于生成专门针对对比预训练的大规模合成数据。
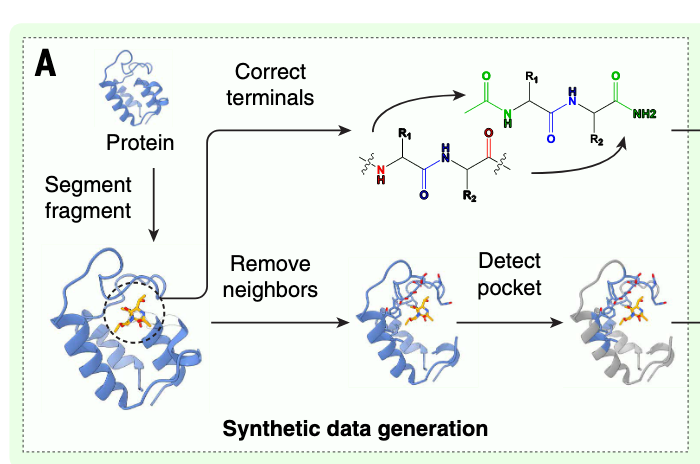
在这种方法中,从仅蛋白质结构中提取短肽片段作为伪配体,其周围区域被指定为伪口袋。蛋白内相互作用与蛋白配体相互作用有许多相似特征,包括氢键、离子吸引、π-π堆叠及其他非共价相互作用(见图S1)。在先前关于配体结合蛋白设计的研究中,蛋白内包装也被用来确定化学基团相对于接触残基骨架的统计优选取向,用于蛋白质-配体界面建模(27)。这一原则是ProFSA发展的基础。为进一步提升模型性能,我们仔细校准了伪配体和伪结合口袋的化学性质分布,使其与真实复合物中观察到的非常匹配(见图S2和S3),从而最大限度地减少合成数据与现实世界数据之间的分布差距。技术细节详见材料与方法的"口袋编码器预训练"部分。将ProFSA框架应用于蛋白质数据库(PDB)数据库(28),获得了550万对伪口袋配体,以促进预训练。训练好的口袋编码器在多个下游任务中进行了评估,如口袋属性预测(表S1)、口袋匹配(表S2)和蛋白质-配体亲和力预测(表S3)。实验结果表明,我们的预训练口袋编码器即使在零输出环境下表现优异,优于许多基于监督的学习模型以及物理和知识模型。这些结果强调了预训练阶段在获得有意义口袋表示方面的成功。关于这些评估的详细信息,见补充文本中的"用ProFSA基准化口袋预训练性能"部分。
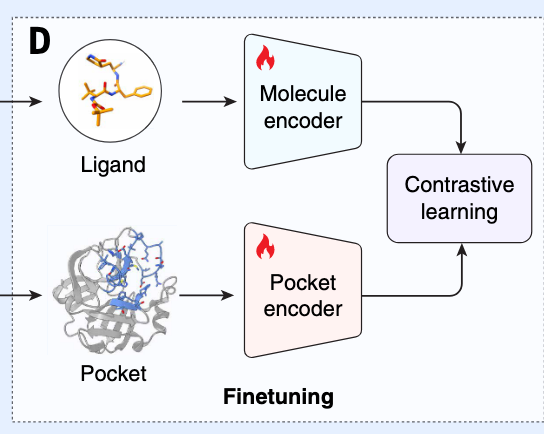
预训练后,分子和口袋编码器进一步微调(见图1D),利用BioLip2数据库收集的4万个实验确定的蛋白质-配体复合结构(29)。鉴于分子的结合构象未知,虚拟筛选仅提供其拓扑结构,我们采用RDKit(30)生成构象,实现了随机构象抽样策略进行数据补充。这种增强使DrugCLIP能够在更准确反映真实世界筛查变异性的数据上训练,从而提升模型的性能和泛化能力。
在筛选过程中(见图1E),我们首先使用训练有素的编码器将分子和口袋表示为载体。随后计算口袋嵌入与分子嵌入之间的余弦相似度,并根据这些相似度评分对候选分子进行排名。排名结果直接基于计算机基准进行评估,DrugCLIP的虚拟筛查结果则通过聚类、对接和人体评估进一步完善,随后进入湿实验室实验(见图1E)。由于分子表示可以离线计算,DrugCLIP筛查效率极高,只需计算简单的余弦相似度并进行后续排名。因此,通过适当的预编码和并行化,DrugCLIP可以用单个GPU加速器评估万亿级靶分子对,这比分子对接等传统计算方法快10,000,000倍以上。