先说 ClinVar 是什么:ClinVar 是 NCBI 的一个变异数据库,核心是把基因变异(variant)和它们的临床意义/疾病关系整理出来,比如某个变异是否致病、和什么疾病相关、证据来自谁提交等。这个下载页就是让你批量拿这些数据。
下载地址位于:
https://www.ncbi.nlm.nih.gov/clinvar/docs/downloads/
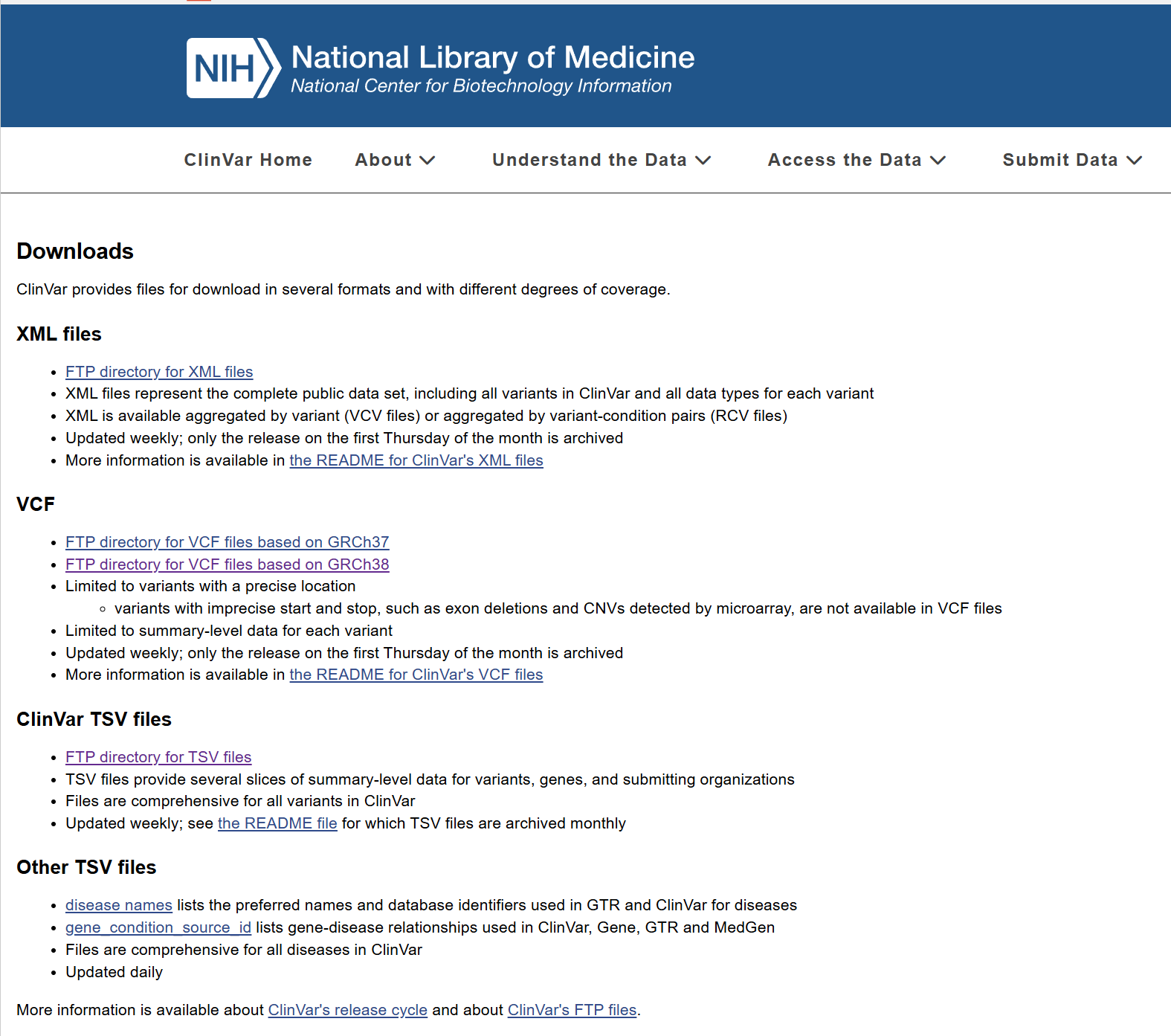
这个页面本质上是在告诉你:ClinVar 提供了几种不同"打包方式"的下载文件 ,它们不是不同的数据集,而是同一套 ClinVar 公共数据的不同格式和粒度。页面把下载分成 4 大类:XML、VCF、ClinVar TSV、Other TSV。
数据格式
1. XML files 是什么
XML 是 最全、最原始、信息最丰富 的版本。页面写得很明确:XML 包含 ClinVar 里的全部公开变异 以及每个变异的所有数据类型。它还有两种聚合方式:
-
VCV(Variant ClinVar) :一个变异为中心
例如"这个具体变异总体上有哪些提交、有哪些解释、关联哪些 condition"。
-
RCV(Reference ClinVar / variant-condition pair) :一个"变异 + 某个疾病"组合为中心
例如"这个变异对于乳腺癌是什么解释,对于另一种病又是什么解释"。
2. VCF 是什么
VCF 是基因组领域最常见的变异格式,适合和你的变异分析流程对接。ClinVar 提供了:
-
基于 GRCh37 的 VCF
-
基于 GRCh38 的 VCF
但这个页面特别提醒了两个限制:
-
只包含有精确基因组位置的变异
-
只包含**summary-level(摘要级)**数据,而不是 XML 那种全细节数据
也就是说,像一些起止位置不精确 的变异,比如某些外显子缺失、微阵列检测到的 CNV,这些不会出现在 VCF 里。
3. ClinVar TSV files 是什么
TSV 就是制表符分隔的文本表格,最适合直接用 pandas、Excel、R 之类读取。页面说它提供的是summary-level 的多个切片 ,覆盖variants、genes、submitting organizations 等信息,而且对 ClinVar 全部变异都是全面的。
也就是说,TSV 更像是把 ClinVar 数据整理成几张比较容易用的表:
-
某张表偏变异摘要
-
某张表偏基因层面
-
某张表偏提交机构层面
所以:
如果你是做统计、筛选、建表、机器学习前处理,TSV 往往最好用。
它比 XML 好处理得多,比 VCF 又更"表格化"。
VCF38的数据下载
https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/
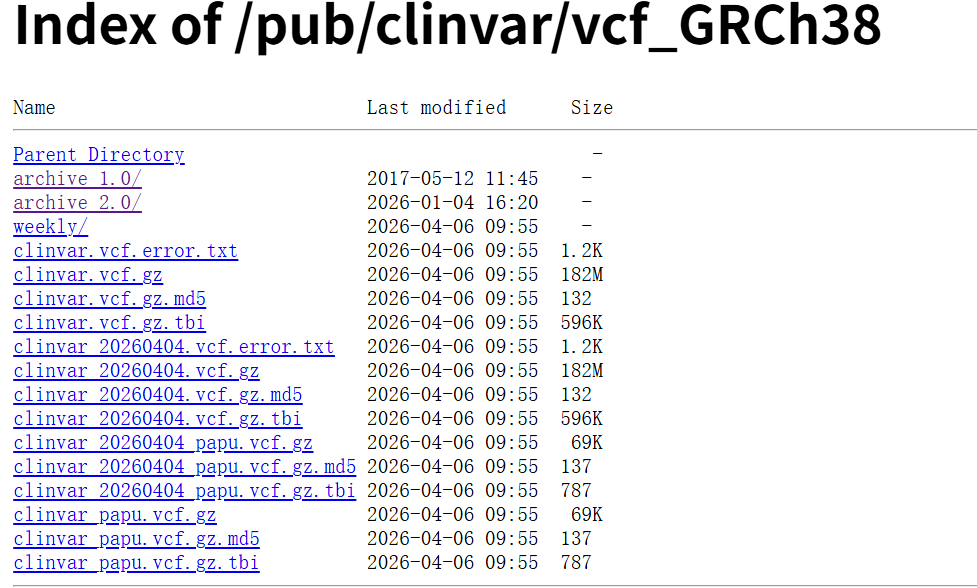
-
archive_1.0/
老版本归档目录。
-
archive_2.0/
新版归档目录。ClinVar 现在每周更新,但通常只把每月第一个周四的版本长期归档。
-
weekly/
每周更新版本的目录,适合想拿最新数据的人。
主 VCF 文件
-
clinvar.vcf.gz
这是当前最新的 GRCh38 主 VCF 文件 。
.vcf.gz表示这是经过 gzip 压缩的 VCF。如果你只是想下载 ClinVar 的 hg38 变异,一般下这个就行。(NCBI FTP)
-
clinvar_20260404.vcf.gz
这是带日期的那个版本快照 ,表示这份文件对应
2026-04-04这一期发布。一般来说,它和同目录里的
clinvar.vcf.gz在这一周通常是同一个内容,只是一个是"固定日期名",一个是"当前最新名"。(NCBI FTP)
md5 校验文件
-
clinvar.vcf.gz.md5
-
clinvar_20260404.vcf.gz.md5
这两个是 MD5 校验值 ,用来检查你下载的 .vcf.gz 有没有损坏。
例如下载完后可以跑:
md5sum clinvar.vcf.gz
cat clinvar.vcf.gz.md5看看两者是否一致。
tbi 索引文件
-
clinvar.vcf.gz.tbi
-
clinvar_20260404.vcf.gz.tbi
这是给 bgzip 压缩的 VCF 配套的 tabix 索引文件 。
有了它,你就可以按染色体区间快速查,不用把整个 182M 文件全读一遍。比如:
tabix clinvar.vcf.gz chr1:1000000-2000000如果你要用 bcftools view -r、tabix、IGV 等按区域读取,通常都需要这个 .tbi。
error 文件
-
clinvar.vcf.error.txt
-
clinvar_20260404.vcf.error.txt
这是生成该 VCF 时的错误/警告日志 。
通常记录的是某些 ClinVar 记录为什么没能写进 VCF,或者转换时遇到的异常。
因为 ClinVar VCF 本来就不是全量 XML,它只收录simple alleles、长度 <10 kb、且端点精确定位到 GRCh37/GRCh38 的变异;像 haplotype、genotype、位置不精确的 CNV、以及大于 10 kb 的变异都不在 VCF 范围里。
所以这个 error 文件你可以理解成:"有哪些记录在 VCF 化时没法正常放进去,或者需要说明"。
papu 文件是什么
-
clinvar_papu.vcf.gz
-
clinvar_papu.vcf.gz.md5
-
clinvar_papu.vcf.gz.tbi
-
clinvar_20260404_papu.vcf.gz
-
clinvar_20260404_papu.vcf.gz.md5
-
clinvar_20260404_papu.vcf.gz.tbi
这里的 papu 是 NCBI 用的缩写,表示:
-
Patch
-
Alternate
-
PAR
-
Unplaced
也就是那些不在主染色体标准坐标上的补充序列位置 。NCBI 专门说明,带 papu 的 companion files 是为了支持这些非 primary chromosome locations 的临床变异数据
你可以粗略理解为:
-
主文件
clinvar.vcf.gz:放在标准主染色体坐标上的 ClinVar 变异
-
补充文件
clinvar_papu.vcf.gz:放在补丁序列、替代位点、PAR 区、未定位序列上的变异
而且你截图里 papu 文件只有 69K,远比主 VCF 小,说明这部分只是一个补充小集合。
想要固定版本的话,只需要下载一个clinvar_20260404.vcf.gz即可:https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar_20260404.vcf.gz