Genomic and ecological drivers of parallel arid adaptation in tree grapes (Vitaceae)
葡萄科木质藤本葡萄属植物平行干旱适应的基因组与生态驱动机制

摘要
全球干旱化趋势不断加剧,解析植物的适应机制具有重要意义。肉质化是植物抵御干旱的关键演化性状,在不同植物类群中多次独立出现,但调控这一性状的内在基因组驱动机制仍缺乏深入研究。本研究结合基因组学、生态学与形态学证据,对葡萄科白粉藤属(Cyphostemma)木本葡萄的干旱适应特征展开探究。该属物种分布范围极广,从热带雨林延伸至荒漠生境,基因组与表型分化显著。
本研究组装了白粉藤属代表性物种的基因组,发现特定长末端重复反转录转座子(LTR-RT)谱系伴随该属物种的辐射分化大量增殖,致使基因内含子显著扩张,这一现象在真双子叶植物中鲜有报道。内含子区域插入的长末端重复反转录转座子,可提升基因组对结构变异的耐受能力,进而推动肉质化性状的演化。肉质化物种的基因组还因基因间区的长末端重复反转录转座子进一步扩增,这类转座子在干旱及季节性生境中反复展现出演化优势。
本研究阐明了内源长末端重复反转录转座子的动态变化 与外界环境压力 共同塑造植物基因组特征的过程。重要的是,研究表明长末端重复反转录转座子群体的随机动态变化能够提升基因组演化潜能,为植物的适应性演化提供基础。
结果与讨论
时空演化框架
为解析白粉藤属物种的长末端重复反转录转座子(LTR-RT)动态演化规律,本研究基于 151 个类群的单拷贝直系同源基因构建了高可信度系统发育框架(补充数据 1)。系统发育树将该属划分为四个支持度良好的演化支:分化最早的聚伞花序支 分布于非洲大陆;马达加斯加支与亚洲支互为姐妹群,二者共同构成非洲大陆支的姊妹谱系(补充图 1、2)。
尽管各主要演化支间存在核质系统发育冲突与核基因树拓扑不一致现象(补充图 3),但分析结果显示,类群间基因交流水平较低,不完全谱系分选(ILS) 是造成拓扑矛盾的主要原因(补充图 4、5),这表明杂交事件对后续基于系统发育树的分析影响极小。
结合葡萄科 172 个物种及火筒树科 2 个外类群的分子钟核基因估算分化时间(补充图 6、补充数据 2),结果表明:白粉藤属起源于晚白垩世(约 7160 万年前) ;四个主要演化支在气候温暖的始新世发生快速分化,这也与该属普遍存在高水平不完全谱系分选的特征相吻合(补充图 4、6)。肉质化类群直至渐新世才逐步出现,这一时期恰好对应全球气候趋于干旱、寒冷的转变阶段<sup>22</sup>。
生物地理重建结果显示,白粉藤属冠群起源于热带非洲。但受早期类群快速辐射分化与大量灭绝事件影响,该属四大演化支分化过程中的扩散历史已难以追溯,古生物地理信号被严重掩盖(补充图 7、补充数据 3)<sup>18</sup>。
LTR-RT 驱动基因组与内含子扩张
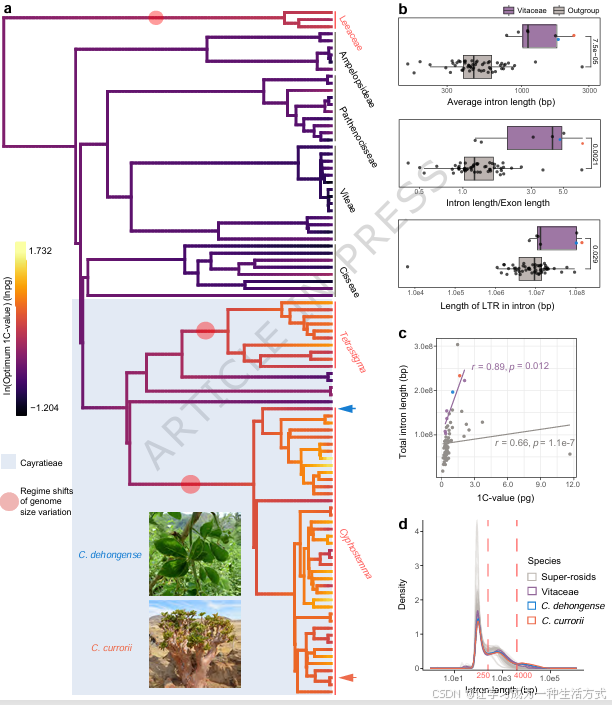
本研究整合已发表文献<sup>10,23,24</sup>与实测数据,共收集葡萄科及外类群 139 组基因组大小数据(补充数据 4),并将数据映射至经过类群精简的葡萄科系统发育树<sup>18</sup>,从宏观系统发育背景解析基因组大小的分化规律。祖先状态重建结果表明,同为葡萄科崖爬藤族的白粉藤属 与崖爬藤属 物种普遍拥有更大的基因组,二者也是葡萄科物种多样性最高的两个属(图 1a)。
本研究采用拟合度最优的奥恩斯坦--刘维尔(OU)模型 分析基因组大小的适应性演化(补充数据 5),该模型认为谱系会在选择压力作用下,使基因组大小趋向某一最优值。结果显示,白粉藤属与崖爬藤属中基因组偏大的谱系,在物种分化早期便出现基因组扩增趋势,且不同谱系的基因组大小逐步趋向各自的最优值(图 1a、补充图 8)。
对 55 个超蔷薇类外类群物种(染色体水平基因组)与 7 个葡萄科物种开展比较基因组分析,发现葡萄科物种的内含子长度出现异常扩张 (图 1b、补充数据 6)。基因组总内含子长度与基因组大小呈显著正相关,证明内含子扩张是推动基因组膨大的重要因素(图 1c)。
在超蔷薇类植物中,内含子长度分布整体呈现三峰特征,在约 250 bp 与 4000 bp 处存在明显谷值(图 1d)。相较于其他超蔷薇类物种,葡萄科(尤其白粉藤属)含有超长内含子(>4000 bp) 的基因数量显著更多,短内含子(≤250 bp)基因占比更低(图 1d)。同时,葡萄科物种(尤以崖爬藤族为代表)的平均内含子长度、内含子 / 外显子长度比值均显著高于其他超蔷薇类物种(图 1b、补充数据 6)。协方差分析(ANCOVA)证实,该类群特异性的内含子扩张现象,与内含子区 LTR-RT 插入长度显著相关(p =0.017;p =3.58×10<sup>-9</sup>,补充数据 7)。
共线性分析可通过基因大片段的保守性推断不同类群的基因组结构演化,结果表明:葡萄属与白粉藤属分化之后,两类群均未再发生额外的古老全基因组加倍事件(补充图 9、补充注释 1),说明葡萄科物种仅经历了真双子叶植物共有的γ 三倍化事件 <sup>25</sup>。
综上,LTR-RT 大量增殖而非全基因组加倍 ,是德宏白粉藤与卡鲁白粉藤基因组、内含子扩张的核心驱动力。通常情况下,LTR-RT 多富集于基因间区,真双子叶植物中因 LTR-RT 插入形成 4000 bp 以上超长内含子的现象十分罕见,该类群内含子平均长度与中位长度分别仅为 619.13 bp、523.00 bp<sup>26,27</sup>。崖爬藤族物种的内含子区大量积累 LTR-RT,代表一种特殊且尚未被充分解析的演化模式,该模式利于反转录转座子在内含子区定植扩增。
白粉藤属 LTR-RT 的多样性特征
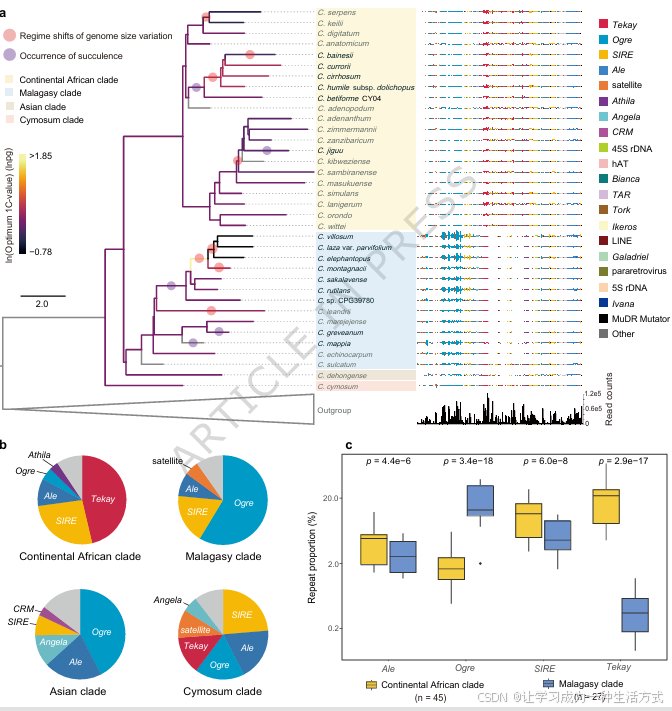
依托系统发育框架(图 2a、补充图 10),本研究利用低深度全基因组测序数据,解析了白粉藤属 74 个物种的重复序列组成(补充数据 8)。结果显示,该属基因组中的 LTR-RT 以少数几个家族为主导(图 2b),证明白粉藤属基因组扩张,主要由少数 LTR-RT 家族的大规模增殖 驱动。
Tekay 、Ogre 、SIRE 是丰度最高的三个 LTR-RT 家族,相较于其他重复序列,这三类转座子是造成该属基因组大小分化的主要原因(图 2a)。不同演化支的 LTR-RT 组成差异显著:非洲大陆支以Tekay 与SIRE 为主,Ogre 占比偏低;马达加斯加支则以Ogre 占绝对优势,SIRE 中等丰度分布,几乎检测不到Tekay 插入(图 2a)。
Ale 为该属第四大 LTR-RT 家族(图 2a、补充数据 8),虽然其在基因组中的整体占比不高,但在白粉藤属所有演化支中均稳定存在(图 2b、c),对基因组扩张起到广泛但相对有限的作用。
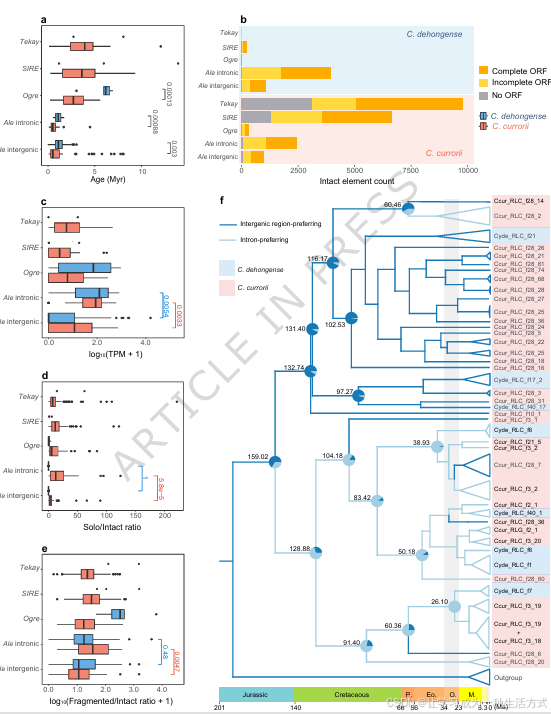
基于德宏白粉藤、卡鲁白粉藤的完整基因组,本研究深入分析上述四类优势 LTR-RT 家族的动态演化,估算其插入时间与消亡速率。相较于非肉质的德宏白粉藤,肉质的卡鲁白粉藤中 Ogre 与 Ale 转座子的插入年代显著更晚(图 3a、补充数据 9--11)。德宏白粉藤中 Tekay 与 SIRE 拷贝数过少,无法开展统计学意义上的插入年代比较,但二者在卡鲁白粉藤中高拷贝的特征,暗示其仍处于近期活跃或持续增殖状态(补充数据 9)。
在卡鲁白粉藤基因组内,Tekay 的消亡速率高于 SIRE ,更新迭代速度更快(补充图 11、补充数据 11)。
甲基化分析显示,卡鲁白粉藤 LTR-RT 整体 CHH 甲基化水平高于德宏白粉藤,但前者基因组内近期活跃的 Tekay 元件反而更多 (图 3a、c,补充图 12a)。通常 CHH 甲基化会抑制 LTR-RT 活性,且该修饰在基因附近的转座子沉默调控中发挥关键作用<sup>28</sup>,二者看似存在矛盾。
究其原因,卡鲁白粉藤的 Tekay 家族中非自主元件占比极高 (开放阅读框不完整或完全缺失,图 3b、补充数据 9),这类元件不易被 CHH 甲基化靶向沉默(补充图 12b)。在整体高 CHH 甲基化的基因组背景下,非自主 Tekay 元件受到的抑制作用较弱,更易存活并不断积累。
这一增殖特征也解释了为何非洲大陆支以 Tekay 为主、马达加斯加支以 Ogre 为主(图 2a、c),反映出两类群截然不同的基因组扩张历史。总体而言,这种演化支特异性的 LTR-RT 动态模式,塑造了白粉藤属物种间差异化的转座子组成(图 2a、补充数据 8)。该特征在属分化早期便已形成并长期保守,与棕榈科及其他诸多植物类群的演化规律相似<sup>29,30</sup>。
内含子区 LTR-RT 的演化动态与起源
在德宏白粉藤与卡鲁白粉藤基因组中,丰度前三的 Tekay 、Ogre 、SIRE 主要造成基因间区扩张 ;而内含子延长则主要由 Ale 元件驱动(补充图 13f)。在两种白粉藤中,Ale 分别占内含子区 LTR-RT 总长度的 58.23%、54.65%(补充图 14、补充数据 12)。Ale 在超长内含子(>4000 bp)中显著富集,是造成两个物种内含子大幅扩张的核心元件(补充图 15)。
相较于 Tekay 、Ogre 、SIRE ,Ale 元件整体插入年代更晚,代表其活性周期更近(图 3a)。转录组数据显示,内含子区的 Ale 元件表达量显著高于基因间区拷贝 (图 3c、补充数据 10--11),这一现象与其基因组位置密切相关:内含子插入会提升转座子与宿主基因共转录的概率<sup>31</sup>。
此外,卡鲁白粉藤中偏好插入内含子的 Ale 元件,其单 LTR / 完整元件比值(SI) 、** 片段化元件 // 完整元件比值(FI)** 显著更高(元件定义详见方法部分)(图 3d、e),说明相较于基因间区的Ale 元件,内含子区的Ale 谱系承受着更强的删除选择压力<sup>32,33</sup>。
该结果表明,基因间区Ale 元件被优先删除,并不能解释Ale 元件在内含子区富集这一现象。较高的删除比率反映出元件更新迭代速度快,这也意味着,相较于Tekay 、SIRE 、Ogre 等其他高丰度 LTR-RT 家族,肉质物种卡鲁白粉藤中内含子区的Ale 元件有更多机会调控基因功能。而在德宏白粉藤中,内含子区与基因间区的Ale 元件在 SI、FI 比值上无显著差异,这可能是由于该物种内多数Ale 谱系仅含单一拷贝所致(补充数据 10)。
本研究结合德宏白粉藤与卡鲁白粉藤的Ale 元件,构建了带分化时间校正的系统发育树,追溯内含子型Ale 谱系的演化起源(图 3f、补充数据 13)。对 GC 含量与元件插入年代开展广义线性模型(GLM)分析,结果显示,随时间推移,甲基化胞嘧啶发生脱氨基作用,逐步转化为胸腺嘧啶,使得碱基替换呈现出时间依赖性特征(补充数据 14),该规律与二穗短柄草中的研究结论一致<sup>34</sup>。
基于上述演化特征,本研究利用逆转录酶基因编码区序列,对新近插入的Ale 元件(插入年代接近 0 百万年)构建时间树;同时将古老元件(插入年代>0.005 百万年)作为 "化石校正点",开展末端定年分析(补充数据 13)。时间树结果显示,内含子型Ale 谱系起源于白垩纪,但其大规模分化发生在渐新世,现存绝大多数高拷贝(拷贝数>50)、偏好插入内含子的Ale 谱系均形成于这一时期(图 3f)。
Ale 元件的分化时间,与白粉藤属物种快速辐射分化、适应不同生境的时间高度吻合(补充图 6),暗示内含子区Ale 元件的大量增殖,推动了该属物种分化过程中的基因组创新。在两个目标物种中,因Ale 插入形成超长内含子的基因,表达量显著降低 ,且所受选择压力更宽松(图 4a、补充图 16)。该结果印证了相关假说:超长内含子会增加转录成本、提升可变剪接出错风险,因此高表达基因通常会筛选淘汰长内含子<sup>35,36</sup>。
肉质化性状的演化
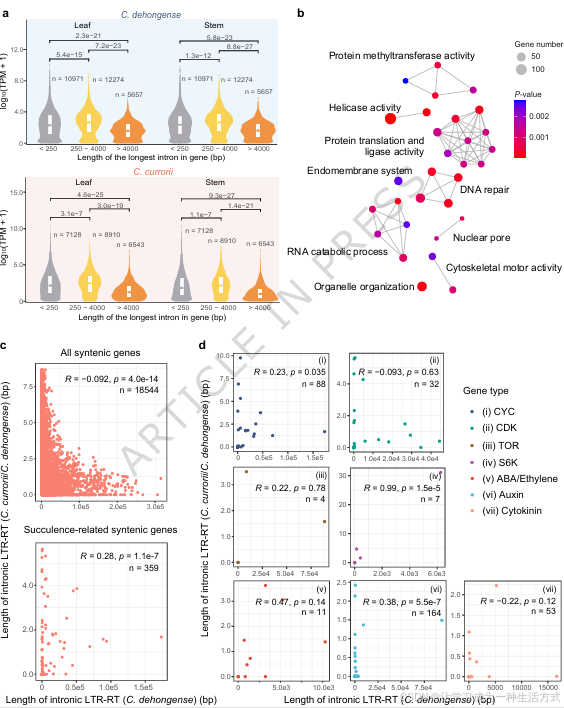
将卡鲁白粉藤中因 LTR-RT 插入形成超长内含子(>4000 bp)的基因,与其在德宏白粉藤中的同源共线基因进行对比分析,发现这类基因显著富集于转录调控、DNA 修复与 RNA 分解代谢 相关通路(图 4b、补充图 17a)。卡鲁白粉藤中此类基因表达量整体偏低,这很可能由基因区更高的 CHG、CHH 甲基化水平所介导(补充图 18)。
已有研究证实,DNA 修复过程中的序列缺失是基因组缩减的重要机制<sup>37</sup>。卡鲁白粉藤体内 DNA 修复相关基因表达下调,会限制基因组序列的丢失;与此同时,Tekay 、Ogre 、SIRE 等高活性 LTR-RT 家族持续扩增、不断积累,最终推动基因组进一步膨大<sup>12,14</sup>。
此外,本研究比对了德宏白粉藤与卡鲁白粉藤全部同源共线基因对的 LTR-RT 插入长度:若某一基因在德宏白粉藤中存在较长的 LTR-RT 插入,其在卡鲁白粉藤中往往出现序列缩短或扩增幅度降低的现象(图 4c、补充图 17)。这反映出植物存在调控机制,可限制内含子过度扩张,以此维持基因正常功能<sup>35</sup>。
但前人研究筛选得到的肉质化发育相关基因 <sup>6</sup>(补充数据 15)并不遵循上述规律。其中,调控细胞大小与维管组织模式建成的细胞周期蛋白、生长素相关基因表现出特殊规律:两个物种的同源基因中,内含子 LTR-RT 插入长度呈显著正相关(图 4d、补充图 17)。该相关性说明,持续的 LTR-RT 插入会对这类基因产生稳定的功能抑制。
与此结论相符,多个肉质化相关基因在长内含子基因集中显著富集(图 4b),其中以生长素与细胞周期蛋白调控的有丝分裂通路基因为主(补充数据 15)。同时,在肉质与非肉质白粉藤物种间表达差异显著的肉质化相关基因,全部集中于生长素、细胞周期蛋白以及核糖体 S6 激酶(S6K)家族,这类基因均是细胞周期调控的关键因子(补充数据 16)。
生长素、细胞周期蛋白与 S6K 基因活性降低,会促使细胞发生核内再复制 ,细胞 DNA 含量升高,最终导致细胞体积增大,这一特征在白粉藤属肉质类群中普遍存在<sup>38-41</sup>。细胞 DNA 含量升高还与气孔密度降低显著关联<sup>42,43</sup>,而气孔密度下降是肉质植物适应干旱环境、提升保水能力的典型特征<sup>2,44</sup>。另有研究表明,生长素相关基因表达受抑,还会调控维管组织的形成与空间分布,对植物水分利用效率起到关键作用<sup>6</sup>。
本研究利用扫描电子显微镜,观察了 22 个白粉藤属物种及外类群物种的叶片表皮结构,共划分出四种叶片表皮类型(补充图 19、补充注释 2)。该结果进一步证实,肉质化性状在白粉藤属中经历了多次独立演化 ,同时明确了肉质物种与非肉质物种在形态上的整体差异。
相较于 9 个非肉质物种,13 个肉质物种的叶片远轴面气孔密度显著更低,且气孔大小变异幅度更大(补充图 20a、补充数据 17)。但上述叶片特征与基因组大小并无显著相关性(补充图 20b),说明气孔特征主要受其他因素调控,例如组织特异性核内再复制引发的 DNA 含量变化、外界环境因子等<sup>45,46</sup>。
值得注意的是,在各演化支 LTR-RT 家族组成整体保持稳定的前提下,肉质化性状仍多次独立起源,该演化模式在其他植物类群中也有报道<sup>29,30</sup>。这表明,肉质化的演化并不需要整个基因组的转座子组成发生重大改变;LTR-RT 插入对特定肉质化相关基因的调控作用 (如改变甲基化水平、影响转录过程),就足以驱动白粉藤属内肉质化性状反复独立演化。
干旱生境对大基因组的选择作用
此前一项宏观演化研究发现,干旱生境中的白粉藤属肉质物种普遍拥有更大的基因组,且该规律在整个真双子叶植物中同样适用<sup>10</sup>。本研究在此基础上,纳入温度、季节性等更多环境因子(补充数据 18),结合白粉藤属物种的重复序列组成开展精细分析,解析基因组大小分化与适应性性状演化背后的分子机制。
首先基于时间校正系统发育树,梳理白粉藤属基因组扩张 与肉质化性状 出现的先后顺序。基因组大小祖先状态重建结果显示,72.8% 的肉质物种在演化过程中,经历了向大基因组方向的选择转变(图 2a、补充图 10)。但不同演化支的演化趋势存在差异:非洲大陆支整体呈现基因组持续扩张;而马达加斯加支基因组在分化初期快速增大,随后又明显缩减(图 2a、补充图 10)。
关键的是,多数类群中肉质化性状的出现早于基因组大小的定向转变 ,这说明基因组大幅扩张并非肉质化演化的必要前提(图 2a)。随机性状关联分析显示,肉质化性状的演化与物种向干旱生境的迁移密切相关(补充图 21),由此推断,干旱生境带来的全新选择压力,驱动了白粉藤属肉质类群的基因组扩张。
鉴于各演化支在基因组大小演化模式、LTR-RT 组成上存在明显分化,本研究采用系统发育广义最小二乘回归(PGLS) ,分别分析每个演化支内环境因子与基因组大小、LTR-RT 占比的关联。分析所用环境变量均基于物种分布数据筛选,取自气候数据库且变量间无相关性(补充图 22、补充数据 18--21)。
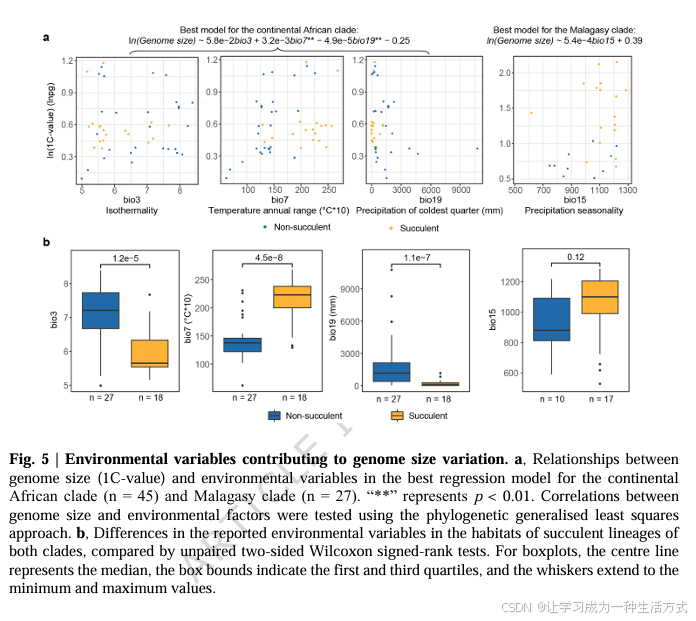
对于非洲大陆支,基因组大小与年温差(生物气候因子 7)呈显著正相关,与最冷季度降水量(生物气候因子 19)呈显著负相关(图 5a、补充数据 19);最优模型中还包含等温性(生物气候因子 3),该因子与基因组大小呈正相关,但相关性不显著。上述特征与非洲大陆支肉质物种所处生境高度吻合:该类群栖息环境干旱、气候季节性强,气温波动大、冷季降水稀少,这类生境特征是驱动其基因组大小发生定向改变的重要因素(图 5b)。
针对马达加斯加支,最优模型分析显示,降水季节性(生物气候因子 15)与基因组大小呈正相关,但未达到显著水平(图 5a、补充数据 19)。同时,该岛屿上的肉质物种与非肉质物种,在降水季节性上也无显著差异(图 5b)。
为探究环境因子是否通过特定长末端重复反转录转座子(LTR-RT)家族的增殖推动基因组扩张,本研究将与基因组大小显著相关的环境变量,分别与Tekay 、SIRE 、Ogre 、Ale 这四类优势 LTR-RT 家族开展系统发育广义最小二乘(PGLS)回归分析(补充数据 22--24)。结果显示,无论是非洲大陆支还是马达加斯加支,所检测的 LTR-RT 家族丰度与基因组扩张均不存在显著线性关联(补充数据 22)。
在非洲大陆支中,年温差与Tekay 元件占比呈负相关(补充数据 25);而马达加斯加支内,各 LTR-RT 家族占比与环境因子之间均未检测到显著关联(补充数据 26)。综合来看,现有结果难以证实环境因子会通过选择性扩增特定 LTR-RT 家族来驱动基因组扩张。相反,本研究表明,环境选择压力主要作用于整体基因组大小 ,而非单一 LTR-RT 组分的构成。
本研究进一步分析了基因组大小与局域精细化环境因子的关联<sup>47</sup>,发现土壤氮含量是两个演化支共同的主导影响因子,但二者响应模式存在差异(补充图 23、补充数据 27--28)。非洲大陆支的基因组大小对不同土层的氮含量表现出差异化响应;马达加斯加支的最优模型显示,基因组大小与表层土壤(0--5 厘米)氮含量呈负相关,但该相关性不显著,这与该岛屿干旱生境异质性高、物种多为浅根系的特征相符。
尽管马达加斯加支整体呈现基因组缩减趋势(图 2a、补充图 10),但在当前氮素匮乏的环境下,其基因组进一步缩小的能力十分有限。这一矛盾现象提示,后续需开展更广范围的比较研究,解析生长于养分受限环境中的植物类群,其基因组约束与生态压力之间复杂的互作关系。
总结
本研究整合基因组、生态与表型数据,阐释了作为基因组大小分化核心机制的 LTR-RT 增殖,如何参与白粉藤属肉质化性状的演化,并解析了该性状与干旱生境的内在关联(补充图 24)。通过对比非肉质物种德宏白粉藤与肉质物种卡鲁白粉藤的基因组,本研究证实,以往鲜有探究的内含子区 LTR-RT 插入 能够提升基因组演化潜能,进而推动肉质化性状形成。
目前 LTR-RT 调控肉质化的具体通路仍有待解析,但本研究筛选出生长素、细胞周期蛋白及核糖体 S6 激酶相关基因,可作为后续功能验证的候选靶点。白粉藤属的基因组分化与肉质性状,既可能在干旱生境中逐步演化形成,也可能是祖先类群在湿润生境中形成的前适应性状 。若是后者,当全球气候趋于干旱时,具备肉质特征的物种便可依托这类预存性状提升生存能力。
归根结底,LTR-RT 在基因间区与内含子区的大量累积引发基因组大幅扩张,同时转座子对基因组功能与基因表达产生调控作用,这极大提升了白粉藤属类群在干旱环境中的适应能力与演化存续能力。
LTR-RT 曾被视作 "垃圾 DNA",如今学界愈发认识到,活跃的 LTR-RT 是植物演化出新适应性性状的动态基因库;类群定向的适应性演化,也可源于 LTR-RT 群体的随机演化过程。白粉藤属内多次独立形成相似适应性性状的现象,体现了 LTR-RT 主导下演化路径的灵活性,这对于全球干旱化背景下的植物适应生存具有重要意义。
未来可结合精细化环境梯度,并扩大取样范围,选取非洲大陆与马达加斯加地区更多肉质、非肉质近缘类群开展比较基因组研究,进一步阐明基因组结构如何介导植物对生态压力的适应性响应。
材料与方法
物种取样与 DNA 测序
本研究采集了葡萄瓮属 118 个物种共 124 份材料的浅层全基因组测序(WGS)数据;另选取葡萄科 54 个近缘物种、火筒树科 2 个物种作为外类群,用于生物地理及重复序列组成分析。
马达加斯加实验材料依据该国环境与可持续发展部出具的科研许可(编号:N_025/23/MEDD/SG/DGGE/DAPRNE/SCBE.Re)采集;肯尼亚实验材料遵循当地环境、气候变化与林业部许可(编号:RESEA/1/KFS 98、RESEA/1/KFS 22)完成取样。所有采样工作均遵守当地法律法规。其余试验材料均取自已有种质库,无需额外采集许可。
采用改良 CTAB 法,从 115 份硅胶干燥叶片样本(包含葡萄瓮属 109 个物种、11 个外类群物种)中提取基因组 DNA。利用 Illumina HiSeq2500 平台开展测序,每个样本产出 6~10 Gb、读长 150 bp 的双端测序数据。同时,从美国国家生物技术信息中心(NCBI)序列读取档案库(SRA)下载 9 个葡萄瓮属物种及 43 个外类群物种的全基因组测序数据。
在开展德宏葡萄瓮 与库氏葡萄瓮 基因组正式测序前,先完成基因组特征评估(基因组大小、杂合率、重复序列占比)。两种植物栽培于国家植物园温室,取新鲜幼叶为实验材料,使用凯杰基因组 DNA 提取试剂盒,按照厂家标准流程提取 DNA。通过 1% 琼脂糖凝胶电泳检测 DNA 是否降解、有无污染,利用 Qubit 4.0 荧光定量仪(美国赛默飞)测定 DNA 浓度。
将两个物种的 DNA 片段化至约 350 bp,经末端修复、加 A 尾、接头连接后构建测序文库。使用 fastp v0.23.2 软件(参数:-average_qual 15 -l 150 -w 6)过滤原始读段,去除低质量碱基与接头序列。基于质控后的 clean reads,采用 Jellyfish v2.2.6 统计 k-mer,并结合 GenomeScope 2.0 评估基因组特征。
基因组调研结果显示,两个物种杂合度均较低、重复序列占比偏高:德宏葡萄瓮基因组大小约 1.1 Gb,杂合率 0.47%,重复序列占比 67.8%;库氏葡萄瓮基因组大小约 1.7 Gb,杂合率 0.63%,重复序列占比 75.1%。该结果为后续长读长测序及基因组组装提供依据。
针对德宏葡萄瓮,构建片段长度≥15 kb 的 PacBio SMRTbell 文库,在 PacBio Sequel 平台完成测序,借助 SMRT Link v6.0 软件产出 HiFi 高精度读段。针对库氏葡萄瓮,每份样本取 3~4 μg DNA 构建牛津纳米孔(ONT)文库,利用 PromethION 平台测序;通过 Guppy v5.0.13 碱基识别软件将原始 fast5 文件转换为 fastq 格式,使用 NanoPlot v1.38.13 评估测序质量。
参照 Rao 等人的方法为两个物种构建 Hi-C 文库,并在 Illumina NovaSeq 6000 平台开展 150 bp 双端测序。
转录组测序与拼接组装
取材于国家植物园温室栽培植株:采集德宏葡萄瓮成熟叶片、茎、卷须、幼根,库氏葡萄瓮成熟叶片、茎、花组织,用于基因组注释。为分析肉质性状相关基因的表达差异,另采集 16 种葡萄瓮属植物的成熟叶片(详见补充数据 1)。
采用改良 CTAB 法提取各样品总 RNA,使用 NEB 公司 Illumina 专用 mRNA 文库制备试剂盒,构建插入片段为 150 bp 的 polyA 富集转录组文库。双端 cDNA 文库经 Illumina 4000 平台测序获得原始数据。
运用 Trimmomatic v0.39 软件(参数:SLIDINGWINDOW:4:20 LEADING:20 TRAILING:20 MINLEN:50)对原始读段进行质量修剪,再通过 Bowtie2 v2.4.5 过滤细胞器来源序列。使用 Trinity v2.15.1 默认参数完成转录本拼接,分别利用 TransRate v1.0.1 和 BUSCO v5.2.2 评估组装质量。
借助 TransDecoder v5.5.0 预测基因最长转录本并完成蛋白序列翻译;最后使用 CD-HIT v4.7(相似度阈值 0.99)去除冗余转录本。
基因组大小、形态特征与生境数据整理
本研究从三类渠道收集葡萄科、火筒树科物种的基因组大小数据:①已发表文献;②植物 DNA C 值数据库(7.1 版,2024 年 2 月检索);③本研究新增的流式细胞术检测数据。
流式细胞术检测参照 Galbraith 等人建立的方法开展。取约 1 cm² 干燥叶片剪碎,置于 2 mL 改良 Galbraith 缓冲液中制备细胞核悬液,经 30 μm 尼龙滤膜过滤后,加入终浓度 50 μg/mL 碘化丙啶(PI)与 0.5 μg/mL 核糖核酸酶(RNase)对细胞核染色。采用 BD LSRFortessa 流式细胞分析仪检测,单样本有效检测颗粒数不少于 5000 个,以此估算细胞核 DNA 含量。
由于葡萄科物种基因组大小差异较大,检测前先利用待测样本调试仪器电压并选定参照标准物种。选用的参照物种及对应 1C 值如下:日本晴水稻(0.43 pg)、野生番茄(0.75 pg)、大豆 Williams 82(1.11 pg)、玉米 CE-777(2.72 pg)、辣椒遵辣 1 号(3.42 pg)。确定参照物种后,将待测物种叶片与参照物种叶片混合剪碎制备匀浆上机检测。
首先通过侧向散射光面积(SSC-A)与 PI 荧光面积(PI-A)散点图设门,去除细胞碎片、死细胞及电子噪声;再依据荧光直方图确定待测物种与参照物种的 G1 期峰(附图 S25)。所有有效样本中,两组 G1 峰的变异系数(CV)均低于 5%。按照下述公式计算待测物种绝对 DNA 含量(1C 值): 待测物种 1C 值 =(待测物种 G1 峰位置 ÷ 参照物种 G1 峰位置)× 参照物种 1C 值 每个个体设置 3 次生物学重复,取平均值作为最终基因组大小结果。
若同一物种存在多条流式检测记录,取所有数据平均值;若无多条记录,则采用最新检测结果。生境数据(生境类型、干旱程度)与形态性状数据(肉质化、块茎、卷须、表皮毛)均查阅植物志、标本记录并结合野外调查整理获得。
基因组组装
使用 Nextdenovo v2.4.0 组装牛津纳米孔(ONT)测序数据得到重叠群(contig),参数设置:random_round = 20 minimap2_options_cns = -x ava-ont -t 10 -k17 -w17 nextgraph_options = -a 1。再通过 Nextpolish v1.4.0 对重叠群进行纠错校正,参数:lgs_options = -min_read_len 1k -max_read_len 100k -max_depth 100 lgs_minimap2_options = -x map-ont。
利用 ALLHiC v5.18.2 流程,结合 Hi-C 短读长的染色质互作信息,对校正后的重叠群进行聚类、排序与染色体锚定,参数:--enz DpnII --CLUSTER $chr_num --break N --fill Y。借助 Juicebox v1.13.017 对组装结果进行人工纠错。选用双子叶植物数据库udicotyledons_odb10 ,通过 BUSCO v5.2.2 评估基因组组装完整度。
重复序列注释
使用 TRF v4.09.1 识别串联重复序列。基于拟南芥 RepBase 重复序列库(2018 年 10 月 26 日版本),在默认参数下运行 RepeatMasker v4.1.5,对两个葡萄瓮属基因组进行重复序列比对注释。
联合 RepeatModeler v2.0.5、LTR_FINDER v1.1 及 LTRharvest v1.6.4 的分析结果,构建全长长末端重复反转座子(LTR-RT)数据库,剔除长度小于 100 bp 或空缺碱基(N)占比超 5% 的序列。参照 80-90-100 规则,使用 LTR_retriever 去除冗余序列:CD-HIT 聚类后,序列相似度>80%、比对覆盖最长序列>90% 且长度>100 bp 的序列判定为冗余。
采用 TEsorter v1.4.6 完成剩余 LTR-RT 的家族分类。结合 LTR_retriever v2.9.9 与自主编写的 Perl 脚本,核查元件完整性并优化数据库。 元件分类标准:截短型 LTR-RT :隶属于同一家族、两端 LTR 完整或近完整,但缺失 LTR 侧翼二核苷酸回文基序及元件两侧 5 bp 靶位点重复序列(TSD)等末端结构;单一 LTR :仅保留单个 LTR,且两侧存在二核苷酸回文基序与 TSD;无法归为上述两类的元件,定义为片段化 LTR-RT 。
基因与非编码 RNA 注释
基因预测结合从头预测、同源比对、转录组证据 三种策略,并针对长内含子基因进行专项校验。
- 从头预测:在屏蔽重复序列的支架序列上,使用 AUGUSTUS v3.2.3、Geneid v1.4、Genescan v1.0、GlimmerHMM v3.04 及 SNAP(2013-11-29 版)进行基因预测;
- 同源预测:下载拟南芥、草莓、葡萄参考基因组,通过 GeneWise v2.4.1 开展蛋白序列比对,完成基因模型注释;
- 转录组辅助:使用 Hisat v2.0.4 将不同组织(叶片、茎、根、花、卷须)的 RNA 测序读长比对至基因组,结合 Stringtie v1.3.3 组装转录本,再利用 PASA v2.4.1 预测基因模型。
剔除无转录组支持、无同源序列、无保守结构域或开放阅读框(ORF)不完整的注释结果,最终通过 EvidenceModeler v1.1.1 整合多组预测结果,得到非冗余基因集。
基因功能注释:将基因序列分别比对至瑞士蛋白质数据库(Swiss-Prot)与 NCBI 非冗余蛋白库(NR),E 值阈值设为 1e-5,依据最佳比对结果注释基因功能;运用 InterProScan v5.31 检索 ProDom、PRINTS、Pfam 等公共蛋白数据库,预测蛋白基序与结构域,并据此分配基因本体(GO)条目;结合 KEGG 数据库比对结果,解析基因参与的代谢通路。
非编码 RNA 注释:使用 tRNAscan-SE 预测核糖体 RNA(rRNA);基于 Rfam 数据库,通过 infernal v1.1.3(默认参数)鉴定其余非编码 RNA。
甲基化水平分析
采集温室栽培的德宏葡萄瓮与库氏葡萄瓮新鲜叶片,采用改良 CTAB 法提取基因组 DNA。将高质量 DNA 片段化至 200--400 bp,利用 EZ DNA Methylation-Gold 甲基化试剂盒进行亚硫酸氢盐转化;经纯化、扩增后,使用 Accel-NGS Methyl-Seq 建库试剂盒构建测序文库。
运用 Bismark v0.24.0(参数:-X 700 --dovetail)对亚硫酸氢盐测序原始数据进行质控,并比对至两个物种的参考基因组。以序列转化率为期望概率,通过二项分布检验识别甲基化胞嘧啶,错误发现率(FDR)P <0.05 判定为有效甲基化位点。参照 Schultz 等人的方法,分别统计基因区域与 LTR-RT 区域的平均甲基化水平。
系统发育重建
本研究基于从崖爬藤族、白粉藤族物种中筛选得到的 1367 个单拷贝直系同源基因,重建葡萄瓮属(Cyphostemma )系统发育树。此前已有研究利用葡萄科叶绿体编码序列与 229 个单拷贝核基因构建该属系统发育树,但这些树无法解析葡萄瓮属各大支系间的亲缘关系,且存在核质基因树拓扑冲突,难以搭建可靠的系统发育框架支撑后续分析。因此,本研究整合崖爬藤族、白粉藤族更多单拷贝直系同源基因,构建高支持度的葡萄瓮属系统发育树。
选取本研究前期组装完成的纤花乌蔹莓(Causonis ciliifera )、美丽拟崖爬藤(Pseudocayratia speciosa )转录组数据,以及库氏葡萄瓮、德宏葡萄瓮、三叶崖爬藤、圆叶白粉藤基因组用于直系同源基因筛选;公共数据库下载的基因组材料信息详见补充数据 6。采用 OrthoFinder v2.5.4 开展多序列比对,fasttree v2.1.11 构建基因树,初步得到 6063 个候选单拷贝直系同源基因簇。通过双向 BLASTN v2.14.1 + 过滤,剔除同一物种内存在多匹配、比对 E 值大于 10⁻⁶的基因簇,最终保留 5223 个基因簇。
利用 MAFFT v7.520 的 L-INS-i 算法完成序列比对,trimAl v1.4.rev22 的automated1参数修剪比对结果;去除比对长度小于 450 bp 的基因簇,剩余 4138 个。借助 TreSpEx v1.1 过滤携带误导系统发育信号的基因,筛选标准包含:不支持已发表研究确定拓扑结构的基因簇、IQ-TREE v2.3.4 构建极大似然树后分支长度极端(超过平均分支 4 倍或短于 0.00005 突变单位)、饱和斜率<0.4、决定系数 R²<0.6、LB 得分标准差>39 或 LB 得分上四分位数均值>35;筛选阈值依据对应分布直方图确定。过滤后剩余 3035 个基因簇。
利用 MCScanX v1.0.0,基于库氏葡萄瓮、德宏葡萄瓮、三叶崖爬藤、圆叶白粉藤基因组检验剩余基因的共线性,最终仅保留 1367 个含共线性基因的基因簇,作为下游分析的单拷贝直系同源目标序列。
采用 HybPiper v2.1.6 流程(参数no_stitched_contig),从葡萄瓮属及外类群浅层全基因组测序数据中提取目标单拷贝直系同源基因编码序列;所有序列经 MAFFT(L-INS-i 算法)比对、trimAl 修剪,并人工核查比对结果。利用 IQ-TREE v2.3.4 构建基因树,在 K80、HKY、GTR 模型中筛选最优替换模型,设置 1000 次自举检验。
以 1367 棵基因树为输入文件,使用 ASTRAL v5.7.8 构建多物种溯祖树(MSC 树);参照 Zhang 等人方法,将自举支持度(BS)低于 10% 的基因树节点折叠,减少低支持度分支的干扰。串联建树方面,将所有同源序列拼接,通过 ModelFinder 筛选最优分区方案,基于 IQ-TREE 采用极大似然法构建系统发育树,非连锁分区模型下设置 5000 次超快自举检验。
基因树冲突分析
采用基因树一致因子(gCF)、全节点确定性值(ICA)、四元组抽样分析(QS)量化基因树拓扑冲突。以 MSC 溯祖树为参照树,在 IQ-TREE 中计算各节点 gCF 值;将自举支持度低于 70% 的节点折叠,依托 Phyparts 流程结合 MSC 树与基因树计算各节点 ICA 值,同步统计一致与冲突二分拓扑数量。
为消除稀疏比对、低支持度带来的误导性基因树拓扑干扰,基于 MSC 树开展 100 次重复四元组抽样分析,获得内部分支的四元组一致性(QC)、四元组差异度(QD)、四元组信息度(QI)三类分值。
本研究量化三大核心诱因对基因树冲突的贡献:杂交渐渗、不完全谱系分选(ILS)、基因树构建误差,分析流程参照 Cai 等人研究。为精准识别各大支系冲突来源并降低计算量,选取代表性物种开展分析(详见补充数据 1),以三叶崖爬藤为外类群;利用 IQ-TREE 构建代表物种极大似然基因树、串联树,ASTRAL 构建 MSC 溯祖树,方法同上。通过 IQ-TREE 计算 gCF 值表征基因树冲突程度。
物种间渐渗可通过三元组拓扑频率偏离 MSC 溯祖模型检出。三元组包含三种拓扑:((A,B),C)、((A,C),B)、((B,C),A)。若仅存在不完全谱系分选(可被 MSC 模型校正),两种次要冲突三元组出现频率均等;若存在渐渗,两类次要三元组频率将出现显著失衡。通过网状指数量化各节点渐渗水平,该指数代表全部三元组中存在渐渗信号的三元组占比。
基于自举基因树,使用 ASTRAL 生成自举物种树;借助 R 包 Phybase v1.4,在 MSC 模型下为每棵自举物种树模拟 1367 棵基因树。统计实测与模拟基因树的三元组频率,计算网状指数,定位三元组频率显著失衡的演化节点。
以种群突变参数 θ 表征不完全谱系分选强度:θ 等于分化时间校准极大似然树得到的突变单位分支长度,除以 MSC 溯祖树得到的溯祖单位分支长度。为评估基因树构建误差的影响,基于 MSC 树,使用 Seq-Gen v1.3.4 模拟 1367 条长度 1771 bp(实测基因比对平均长度)的基因序列;采用 RAxML v8.2.12(参数-f b)基于模拟序列构建基因树,统计实测物种树各节点可被基因树正确还原的频次。最后依托 R 包 relaimpo v2.2-6 线性回归模型,估算三类因素对基因树冲突的相对贡献度。
以 MSC 溯祖树为初始树,依托 Julia 包 PhyloNetworks v0.12.0 内置的 SNaQ 四分组物种网状演化算法,检测葡萄瓮属各大支系间潜在基因流;选取最优杂交节点数对应的网状演化树作为初始拓扑,开展 100 次自举重复分析,每组重复执行 100 次 SNaQ 搜索。
生物地理分析
每个葡萄瓮属物种选取 1 份代表材料用于分化时间估算;扩大取样至整个葡萄科(补充数据 1、2),引入可靠校正点提升分化时间校准精度。结合化石与次级时间校准约束葡萄科系统发育分化时间:
- 北美北达科他州晚古新世Ampelocissus parvisemina 种子化石,限定蛇葡萄--葡萄支系冠群年龄为 56.8--6200 万年;
- 英格兰南部伦敦黏土下 Bagshot 层晚始新世Vitis glabra 化石,限定葡萄亚属冠群年龄下限 3400 万年;
- 依据 Magallón 与 Castillo 的次级校准结果,将葡萄科主干起源年龄上限约束为 9170 万年。
采用保守校准策略,设置带软边界的均匀先验分布,先验密度 2.5%。
为减少缺失数据,利用 HybPiper 提取葡萄科已报道的 229 个单拷贝同源基因,通过 ASTRAL 构建系统发育树;借助 SortaDate 筛选 50 个近似分子钟基因,依托 PAML v4.10.7 的 MCMCTree 模块估算分化时间,设置生灭模型、独立替换速率、HKY85 替换模型(α=0.5)。每 1000 代取样 1 次,总计获得 10000 份样本,舍弃前 20% 样本作为老化样本;Tracer v1.7 验证所有参数有效样本量均大于 200,独立运行两次确保结果收敛。
基于修剪后的葡萄科分化时间树(仅保留葡萄瓮属与崖爬藤族外类群)开展祖先分布区重建,将全球分布划分为 6 个区域(附图 7):(A) 非洲热带大陆、(B) 非洲南部、(C) 毛里求斯、(D) 亚洲(含亚洲大陆、印度次大陆、马来群岛)、(E) 马达加斯加、(F) 澳大拉西亚(含澳大利亚大陆、新几内亚)。依托 RASP v4.2 内置 R 包 BioGeoBEARS,分别运行 DEC、DIVALIKE、BAYAREALIKE 三类模型,依据校正赤池信息准则(AICc)筛选最优模型。
基于分化时间树,采用 R 包 phytools v1.0-1 贝叶斯随机性状映射方法重建葡萄瓮属肉质化性状与生境的祖先状态;设置两条 MCMC 链,运行 5000 次迭代,舍弃前 20% 迭代样本。
超蔷薇类植物基因组比较
为解析葡萄科基因组大小、内含子长度的扩张规律,本研究在超蔷薇类大类群背景下开展葡萄科基因组比较。从公共数据库下载代表物种高质量染色体水平长读长组装基因组(补充数据 6),筛选标准:①测序深度>30×(基因组 11.9 Gb 的蚕豆除外);②scaffold N50>5 Mb;③BUSCO 完整度>90%;④基因注释经多组外部证据校正;⑤重复序列注释完整且多重证据支撑。
利用 R 语言lm函数分析基因组大小与总内含子长度、平均内含子长度、内外显子长度比值的相关性;采用与德宏葡萄瓮、库氏葡萄瓮完全一致的流程识别、分类下载基因组中的重复序列。在 R 中执行双侧协方差分析(ANCOVA),检验基因组大小、内含子区长末端反转座子(LTR-RT)长度、演化支系类型对内外显子长度比值的影响。
葡萄科与葡萄瓮属基因组大小演化
依托 R 包 phytools 极大似然法重建葡萄科、葡萄瓮属祖先基因组大小状态。科水平重建时,裁剪本研究前期 495 个物种的叶绿体系统发育树,仅保留具备基因组大小数据的类群,最大化取样数量;葡萄瓮属分析采用基于 1367 个单拷贝同源基因构建的 MSC 溯祖树。
针对两类类群基因组大小演化模式,重点验证两点:①葡萄瓮属分化过程中是否形成支系特异性最优基因组大小;②演化区间转换是否驱动最优基因组大小形成。借助 R 包 OUwie v2.6 筛选最优演化模型:
- BM1:布朗运动基础模型,全局单一随机演化速率 σ²;
- BMS:允许不同支系拥有独立演化速率 σ²;
- OU(奥恩斯坦--乌伦贝克)模型:同时包含向最优值 θ 趋同的适应演化速率 α 与随机速率 σ²;细分 OU1(全局单一最优值 θ)、OUM(支系独立 θ)、OUMV(θ、σ² 均支系特异)、OUMA(θ、α 均支系特异)、OUMVA(θ、α、σ² 全部支系特异)。
针对存在多最优值的模型,区分肉质、非肉质类群设定差异化演化假设;依据校正赤池信息准则,葡萄科最优模型为 OUMVA,葡萄瓮属最优模型为 OUMA(补充数据 5)。
依托 R 包 bayou v2.1 识别葡萄科、葡萄瓮属基因组大小演化的离散区间转换位点。该模型假设每条分支代表的演化支系在演化历史中存在定向选择的性状最优值 θ,θ 由适应参数 α 决定,偏离最优值的随机波动由恒定参数 σ² 描述。独立运行两条 MCMC 链,迭代 50 万次,每 200 代取样一次,舍弃前 20% 迭代样本;通过 bayou 包summary函数校验所有参数有效样本量>200。最后利用plotSimmap.mcmc、plotBranchHeatMap函数分别可视化演化区间转换位点与各分支最优基因组大小,后验概率阈值设为 0.3。
重复序列组成分析
基于 Galaxy 平台 RepeatExplorer2 流程,利用浅层全基因组测序数据分析葡萄瓮属物种重复序列组成(补充数据 1);本研究测序深度区间 0.2--20×,满足该软件 0.1--0.5× 的推荐分析深度。使用 fastq 双端测序预处理工具将读长修剪至 100 bp,剔除低质量读段;借助 seqtk v1.4 为每个物种随机抽取 250 万对 reads,达到软件要求的 0.1--0.5× 基因组覆盖度。
通过 TAREAN 流程基于序列相似性聚类鉴定重复序列,后续人工校正注释;依据基因组大小标准化各类重复序列丰度,使用 RepeatExplorer2 配套 R 脚本plot_comparative_clustering_summary.R绘图。按葡萄瓮属各演化支系分组,统计支系内所有取样物种不同重复序列的平均占比。
长末端重复反转座子(LTR-RT)支系划分
结合系统发育关系、序列聚类相似性与 LTR-RT 序列比对点阵图,完成德宏葡萄瓮与库氏葡萄瓮 LTR-RT 支系鉴定。依据 Seberg 和 Peterson 的观点,同源性 是划分 LTR-RT 不同支系的核心判定依据,因此 LTR-RT 分类需遵循其系统发育拓扑关系。
首先基于 LTR_retriever 构建的非冗余重复序列库,利用反转录酶(RT)氨基酸序列构建极大似然(ML)系统发育树,初步划分 LTR-RT 支系。RT 序列演化速率适中,能更准确反映序列同源关系;建树工具为 IQ-TREE,选用 LG 氨基酸替换模型,设置 5000 次超快自举检验。依据树拓扑划分支系:由于输入为去冗余序列库,多数单一序列独立为一个支系,仅高度聚类的多条序列归为同一支系;非冗余库中缺失 RT 编码区的 LTR-RT 临时各单独作为一支。
随后采用 TEsorter 对完整序列库剩余 LTR-RT 进行家族注释,再通过 BLASTN 比对分配至对应支系;依据 80-90-100 规则无法匹配非冗余库中已知 LTR-RT 的序列,定义为全新支系。将同一支系所有 LTR-RT 序列拼接,借助 Geneious v.2021.2.2 绘制比对点阵图验证支系内序列相似度,将与本支系其余序列分化显著的序列拆分,新建独立支系。
利用 MAFFT(--auto参数)比对同一家族各支系的一致性序列,若两支系一致性序列相似度>80%,则合并为同一支系。截短型、单一 LTR、片段化 LTR-RT 均通过 BLASTN 相似性检索,按照相同 80-90-100 规则归入对应支系。
LTR-RT 特征统计
统计完整型、截短型、单一型 LTR-RT 的多项特征:全长 LTR-RT 长度、两端 LTR 长度、内部序列占比、GC 含量、距基因 / 外显子距离、插入时间、转录表达量。针对完整 LTR-RT,依托 TEsorter 识别开放阅读框(ORF),统计内部各编码蛋白有无:衣壳蛋白(GAG)、天冬氨酸蛋白酶(AP)、整合酶(INT)、反转录酶(RT)、核糖核酸酶 H(RH)。
LTR-RT 插入时间估算
通过两端 LTR 序列的核苷酸分歧度(错配位点)除以基因组突变速率,计算完整 LTR-RT 的插入时间。 使用 LASTZ v.1.04.22(参数T = 2 C = 2 H = 2000 Y = 3400 L = 6000 K = 2200)比对葡萄与两种葡萄瓮代表物种的基因组共线性区段,统计有效多态位点分歧度(剔除 N 碱基与比对空位)。参考 You 等人研究,设定葡萄与两种葡萄瓮分化时间为 7102 万年;最终计算得到德宏葡萄瓮基因组突变速率 7.22×10⁻⁹,库氏葡萄瓮为 3.72×10⁻⁸。
LTR-RT 表达量分析
基于转录组数据统计完整 LTR-RT 转录水平:不同组织 RNA 测序读段通过 Hisat2 v.2.1.0 比对至基因组,samtools v.1.18 重排比对文件;featureCounts v.2.0.6 默认参数统计各 LTR-RT 匹配读段数,自编 Python 脚本计算每百万转录本(TPM)数值。TPM=0 的 LTR-RT 判定为无转录活性,绘图时剔除。采用威尔科克森符号秩检验,比较不同特征 LTR-RT 的表达差异显著性。
针对每个 LTR-RT 支系,计算单一 LTR / 完整 LTR 比值(SI)、片段化 / 完整 LTR 比值,以及各项序列特征均值,全部计算通过自编 Perl 脚本完成;截短、单一、片段化 LTR-RT 详细信息存放于 figshare 数据库。
基因组 LTR-RT 动态演化分析
对完整元件数量>50 的 LTR-RT 家族、支系,绘制插入年代分布与插入速率曲线,借助 R 包 TE v.0.3-0 估算元件消亡速率与半衰期。
已有研究表明,甲基化胞嘧啶脱氨基作用会造成元件年龄与 GC 含量呈负相关。本研究对两种葡萄瓮中完整元件数量>30 的每个家族、支系,检验插入年代与 GC 含量的相关性;在 R 中调用glm构建广义线性模型,绝大多数支系二者呈显著负相关(补充数据 14),证明 LTR-RT 内部胞嘧啶以恒定速率突变为胸腺嘧啶。
依托化石生灭演化模型,对 Ale 类 LTR-RT 支系开展分化时间校正。将插入年代趋近于 0 的元件定义为 "现存元件",其余为 "化石元件"。基于 LTR-RT 极大似然系统发育树,各大演化支选取代表性元件用于时间校正,每个支系最多选取 5 条序列避免采样偏倚;由于完整开放阅读框会影响元件甲基化水平,仅保留含完整 ORF 的序列。
Ale 支系校正数据集包含德宏 / 库氏葡萄瓮 55 个现存元件、27 个化石元件,选取 4 条 Tork 类元件作为外类群(补充数据 13)。采用 MAFFT L-INS-i 算法比对反转录酶氨基酸序列,剔除空位占比>50% 的位点;修剪后的蛋白比对文件依据植物高频密码子回译为核苷酸序列。
使用 BEAST v.2.4.8 构建时间树,采用末端定年策略、GTR+R 替换模型、对数正态松弛分子钟;独立运行两条 MCMC 链,迭代 5 亿代,每 1000 代取样一次。基于该时间树,借助 R 包 phytools 的贝叶斯随机性状映射方法重建内含子偏好祖先状态,设置两条 MCMC 链、5000 次迭代。
LTR-RT 插入对宿主基因的影响
比较德宏、库氏葡萄瓮不同最长内含子长度基因的表达水平。以基因最长内含子长度分组:内含子大幅扩张会引发转录错误、造成基因功能异常,可作为自然选择压力的指示指标。
为验证长内含子基因是否受到松弛选择,利用 PAML v.10.4.7 计算葡萄科 5 个物种(德宏葡萄瓮、库氏葡萄瓮、三叶崖爬藤、圆叶白粉藤、葡萄)及外类群拟南芥直系同源基因的非同义 / 同义替换比值(dN/dS);在 R 中调用cor.test检验葡萄瓮属 dN/dS 与内含子长度的相关性。结合 MCScanX 鉴定的共线性基因,对比两种葡萄瓮共线性基因内 LTR-RT 插入长度,解析内含子扩张模式,重点关注文献报道中与肉质性状发育相关的基因。
探究超长内含子功能效应:筛选库氏葡萄瓮中因 LTR-RT 插入形成超长内含子(>4000 bp)、且相较于德宏葡萄瓮显著扩张的基因,开展 GO 功能富集分析;以库氏葡萄瓮全部注释基因为背景集,R 包 clusterProfiler v.4.4.4 完成富集,FDR 校正后p <0.05 的条目判定为显著富集。
利用 18 个葡萄瓮属类群转录组(补充数据 1),对比肉质 / 非肉质物种基因表达差异;R 包 edgeR v.3.38.4 筛选差异基因,筛选标准:FDR p ≤0.05、|log₂倍数变化 |≥1。
葡萄瓮属叶片表皮解剖特征
选取具备基因组大小数据的代表性肉质、非肉质葡萄瓮物种,扫描电镜(SEM)观测叶片表皮特征;以外类群三叶崖爬藤、轮叶白粉藤作为对照。参考已有研究,以保卫细胞长度代表气孔尺寸,消除气孔开闭带来的测量误差。
扫描电镜 200 倍放大拍摄叶片中部上表皮、下表皮图像,统计细胞大小与气孔密度;每个物种 3 份生物学重复,每侧表皮随机选取 5 个视野。ImageJ v.1.54f 测量气孔数量、保卫细胞长度、气孔密度,气孔指数计算公式: \(\text{气孔指数}(\%)=\frac{\text{气孔数}(S)}{\text{气孔数}(S)+\text{表皮细胞数}(E)} \times 100\) 同时计算保卫细胞长度均值与标准差;依据表皮形态特征将叶片表皮划分为 4 类(详见补充注释 2)。
环境因子关联分析
整合全球生物多样性信息库(GBIF)、标本记录与野外调查,整理葡萄瓮属物种分布环境数据。无精确坐标但具备区县以下分布信息的标本,结合物种生境特征在谷歌地球手动匹配坐标;R 包 coordinatecleaner v.2.0-20 清洗坐标,并参照世界植物在线数据库(POWO)人工核对。
从 CHELSA v2.1、glUV、全球干旱度与潜在蒸散数据库(30 角秒分辨率)下载 36 项环境变量;依据物种分布位点提取对应环境因子,计算每个物种均值,剔除种间变异极小的变量。同时从 SoilGrids250m 2.0 获取 0--5 cm、5--15 cm、15--30 cm 土层全氮数据,引用已发表数据集获取全球有效磷含量,提取海拔作为影响基因组大小的局地环境因子。
基于葡萄瓮属 MSC 溯祖树,采用系统发育广义最小二乘回归(PGLS)分析基因组大小与环境因子的关联。由于非洲大陆支系与马达加斯加支系基因组大小演化规律存在差异,两类群分开分析。
R 包 corrplot v.0.92 评估环境因子多重共线性,每组高度相关变量仅保留对基因组大小解释力最优的一项;R 包 caper v.1.0.3 遍历所有环境因子组合构建 PGLS 模型,依据校正赤池信息准则(AICc)筛选最优模型。同步对土壤氮、磷、海拔开展相同 PGLS 回归分析。
为验证 LTR-RT 扩增是否驱动基因组扩张,采用 PGLS 回归分析基因组大小与 LTR-RT 总占比的相关性;仅选取 4 种平均占基因组比例>3%、可造成明显基因组波动的高丰度 LTR-RT 家族:Ale、Ogre、SIRE、Tekay,遍历四者所有组合作为预测变量构建回归模型。
进一步探究 LTR-RT 扩张是否与环境(尤其干旱程度)相关,采用 PGLS 分析 4 类高丰度 LTR-RT 占比、最优环境模型变量、全球干旱指数三者间关联,纳入全部协变量检验。
统计分析
转录组、亚硫酸氢盐甲基化测序每组设置 3 次生物学重复。所有组间差异比较采用 R 中无配对双侧威尔科克森秩和检验,p <0.05 判定差异显著;变量相关性采用斯皮尔曼秩相关检验,调用 R 函数cor.test。图表旁标注对应统计样本量。
数据可用性
德宏葡萄瓮、库氏葡萄瓮基因组、转录组测序原始数据与基因组组装序列已存储于国家基因组科学数据中心(NGDC),生物项目编号 PRJCA058853,访问链接:https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA058853。ONT、PacBio、Hi-C 测序数据登录号见补充数据 29,基因组组装序列登录号见补充数据 6,转录组数据登录号见补充数据 1。
本研究产生的葡萄瓮属及外类群浅层全基因组测序数据存放于美国国家生物技术信息中心(NCBI),生物项目编号 PRJNA1274375,各 SRA 测序文件登录号详见补充数据 1、2。文中用到的基因组大小数据集、环境变量文件全部收录于补充材料;比较基因组分析所用超蔷薇类物种基因组组装与注释登录号见补充数据 6。
LTR-RT 元件详细注释、葡萄瓮物种地理分布、重复序列与基因注释、甲基化测序数据、叶片扫描电镜原始图像均存放于 figshare 数据库,DOI 链接:https://figshare.com/s/71cf4e8d081c59fe52d7。用于绘制箱线图、小提琴图、散点图、柱状图、折线图及表格均值统计的全部原始数据与代码同步上传至该数据库。