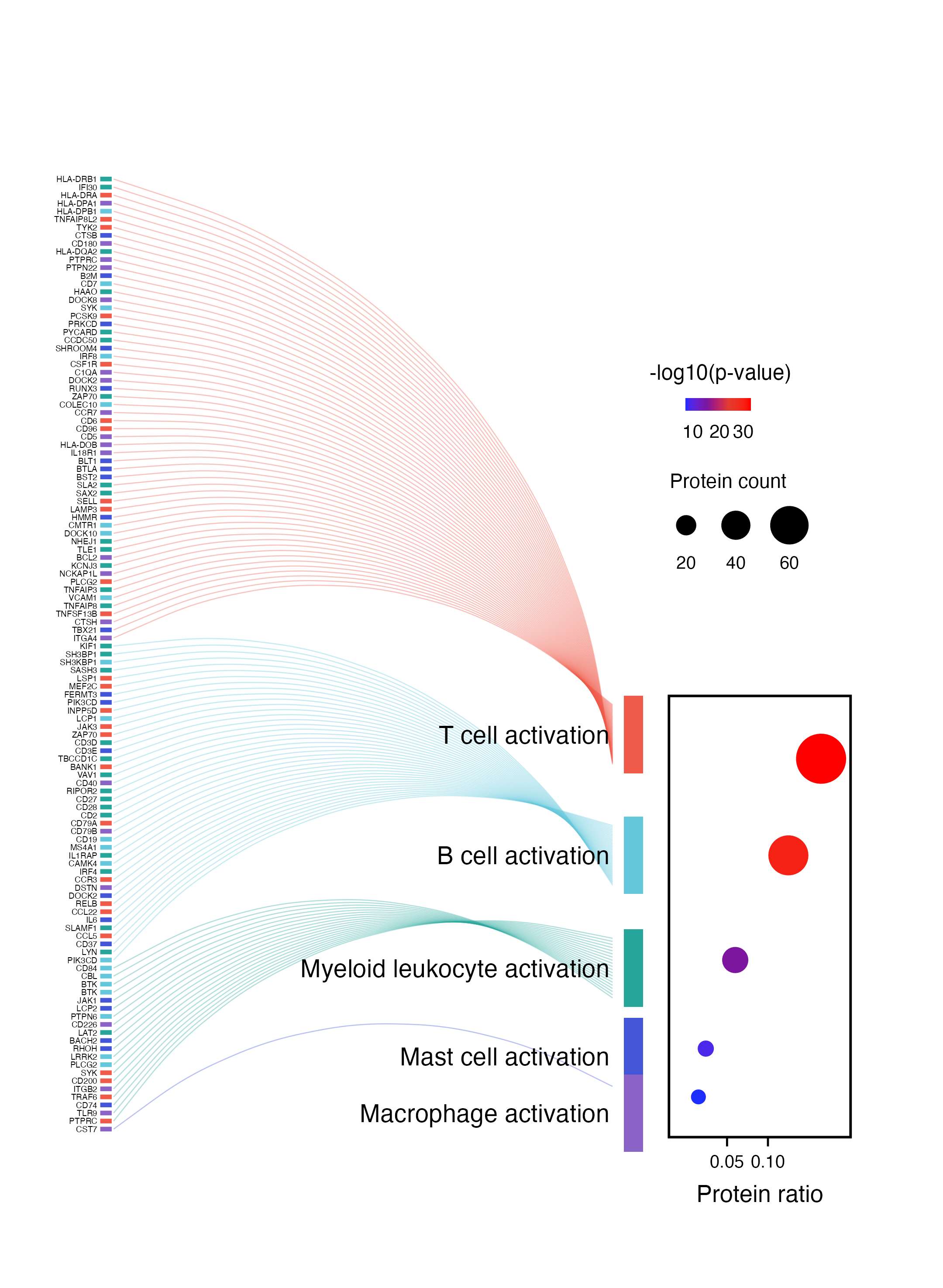
单细胞语境下的"蛋白/基因 → 细胞状态"连接图。左侧展示差异蛋白或 marker,弧线表示这些蛋白归入不同免疫细胞激活状态,右侧气泡图再补充每个细胞状态的比例、显著性和命中数量。
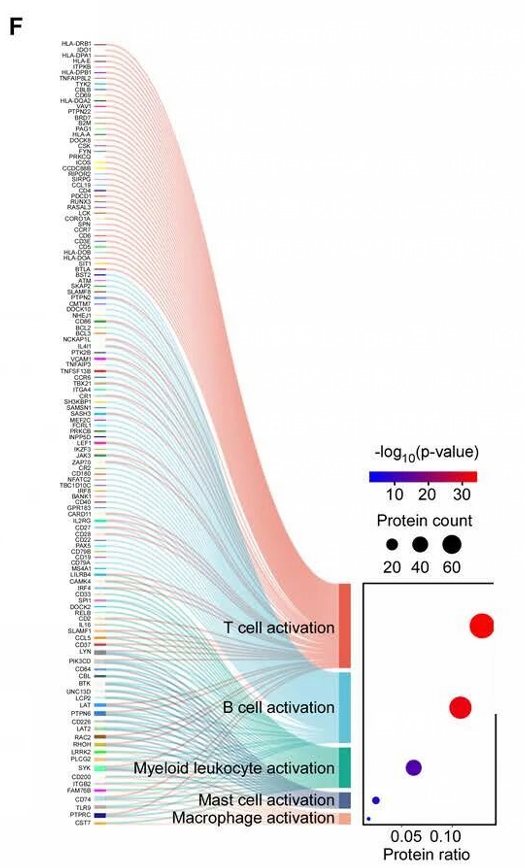
图片来源
| 项目 | 内容 |
|---|---|
| 文章 | Spatial multi-omics implicate the interaction between Tpex and B cells in tertiary lymphoid structures after neoadjuvant therapy |
| 期刊/年份 | Cancer Discovery,2026 OnlineFirst |
| 图号 | 原文截图面板 F |
| DOI/链接 | https://doi.org/10.1158/2159-8290.CD-25-0806 |
这篇文章聚焦新辅助治疗后三级淋巴结构中 Tpex 与 B 细胞互作。截图这个 panel 用蛋白/基因集合连接到不同免疫细胞激活状态,例如 T cell activation、B cell activation、Myeloid leukocyte activation 等。
图片解读
这类图可以拆成三层:
- 左侧蛋白/基因列表:每一行是一个蛋白或基因,旁边短色条可以标记来源、分组或蛋白类别。
- 中间弧线流向:每条弧线表示该蛋白被归入某个单细胞免疫状态。
- 右侧气泡图:横轴是 Protein ratio,颜色表示
-log10(p-value),点大小表示 Protein count。
这里的核心表达是:哪些蛋白集合更集中地指向 T 细胞、B 细胞、髓系细胞、肥大细胞或巨噬细胞相关激活状态。
输入数据
建议准备两张输入表。
1. 蛋白-细胞状态连接表:protein_cellstate_links.csv
| 列名 | 含义 |
|---|---|
protein_label |
左侧展示的蛋白或基因名 |
gene_y |
蛋白在纵轴上的排列位置 |
cell_state |
蛋白对应的单细胞状态 |
cell_state_y_link |
弧线连接到细胞状态色块的位置 |
tick_color |
左侧短色条颜色 |
2. 细胞状态统计表:cellstate_enrichment.csv
| 列名 | 含义 |
|---|---|
cell_state |
细胞状态名称 |
protein_count |
命中蛋白数量 |
protein_ratio |
命中蛋白比例 |
neg_log10_p |
-log10(p-value) |
cell_state_color |
细胞状态颜色 |
cell_state_y |
细胞状态在纵轴上的中心位置 |
r
library(tidyverse)
library(scales)
links <- read_csv("protein_cellstate_links.csv", show_col_types = FALSE)
cell_info <- read_csv("cellstate_enrichment.csv", show_col_types = FALSE)需要示例数据的后台 添加小编 领取,调整好数据结构,以下代码可以直接复制粘贴运行。
第一步:固定细胞状态顺序和颜色
r
cell_levels <- cell_info$cell_state
links <- links |>
mutate(cell_state = factor(cell_state, levels = cell_levels))
cell_info <- cell_info |>
mutate(cell_state = factor(cell_state, levels = cell_levels))
cell_cols <- setNames(cell_info$cell_state_color, cell_info$cell_state)第二步:定义右侧气泡图区域
这里把气泡图作为整体画布中的一个小区域来画。这样更容易还原原图中"弧线图 + 右侧嵌入气泡图"的版式。
r
panel_xmin <- 6.45
panel_xmax <- 8.35
panel_ymin <- 0
panel_ymax <- max(cell_info$cell_state_y + 4.8)
dot_df <- cell_info |>
mutate(
dot_x = rescale(
protein_ratio,
to = c(panel_xmin + 0.18, panel_xmax - 0.18),
from = c(0, 0.18)
),
dot_y = c(47, 35, 22, 11, 5),
dot_col = col_numeric(
palette = c("#1d2cff", "#7c169f", "#e23b2e", "#ff0000"),
domain = c(5, 32)
)(neg_log10_p),
dot_size = rescale(protein_count, to = c(2.4, 10.5), from = c(5, 65))
)第三步:准备图例和细胞状态色块
r
x_ticks <- tibble(
ratio = c(0.05, 0.10),
x = rescale(
ratio,
to = c(panel_xmin + 0.18, panel_xmax - 0.18),
from = c(0, 0.18)
)
)
color_legend <- tibble(
x = seq(panel_xmin + 0.18, panel_xmin + 0.85, length.out = 80),
y = 91,
value = seq(5, 32, length.out = 80)
) |>
mutate(
fill_col = col_numeric(
palette = c("#1d2cff", "#7c169f", "#e23b2e", "#ff0000"),
domain = c(5, 32)
)(value)
)
size_legend <- tibble(
x = c(panel_xmin + 0.18, panel_xmin + 0.70, panel_xmin + 1.26),
y = 76,
count = c(20, 40, 60),
point_size = c(3.7, 5.6, 7.6)
)
cell_bar_df <- cell_info |>
mutate(
xmin = 5.98,
xmax = 6.18,
ymin = cell_state_y - 4.8,
ymax = cell_state_y + 4.8
)第四步:绘制蛋白到细胞状态的弧线
r
p <- ggplot() +
geom_curve(
data = links,
aes(
x = 0.64,
y = gene_y,
xend = 5.86,
yend = cell_state_y_link,
color = cell_state
),
curvature = -0.34,
linewidth = 0.24,
alpha = 0.36
) +
geom_segment(
data = links,
aes(x = 0.50, xend = 0.62, y = gene_y, yend = gene_y, color = tick_color),
linewidth = 1.0,
show.legend = FALSE
) +
geom_text(
data = links,
aes(x = 0.47, y = gene_y, label = protein_label),
hjust = 1,
size = 1.34,
color = "black"
)第五步:添加细胞状态色块和名称
r
p <- p +
geom_rect(
data = cell_bar_df,
aes(xmin = xmin, xmax = xmax, ymin = ymin, ymax = ymax, fill = cell_state),
color = NA,
show.legend = FALSE
) +
geom_text(
data = cell_info,
aes(x = 5.83, y = cell_state_y, label = cell_state),
hjust = 1,
size = 4.0,
color = "black"
)第六步:添加右侧气泡图
r
p <- p +
annotate(
"rect",
xmin = panel_xmin,
xmax = panel_xmax,
ymin = panel_ymin,
ymax = panel_ymax,
fill = NA,
color = "black",
linewidth = 0.55
) +
geom_point(
data = dot_df,
aes(x = dot_x, y = dot_y),
color = dot_df$dot_col,
size = dot_df$dot_size
) +
geom_segment(
data = x_ticks,
aes(x = x, xend = x, y = panel_ymin, yend = panel_ymin - 1.15),
linewidth = 0.45,
color = "black"
) +
geom_text(
data = x_ticks,
aes(x = x, y = panel_ymin - 3.0, label = sprintf("%.2f", ratio)),
size = 2.85,
color = "black"
) +
annotate(
"text",
x = mean(c(panel_xmin, panel_xmax)),
y = panel_ymin - 7.0,
label = "Protein ratio",
size = 3.8
)第七步:添加图例并导出
r
p <- p +
geom_tile(
data = color_legend,
aes(x = x, y = y),
fill = color_legend$fill_col,
width = 0.012,
height = 1.6
) +
annotate("text", x = panel_xmin + 0.54, y = 95.0, label = "-log10(p-value)", size = 3.4) +
annotate("text", x = panel_xmin + 0.25, y = 87.7, label = "10", size = 3.0) +
annotate("text", x = panel_xmin + 0.53, y = 87.7, label = "20", size = 3.0) +
annotate("text", x = panel_xmin + 0.78, y = 87.7, label = "30", size = 3.0) +
annotate("text", x = panel_xmin + 0.62, y = 81.5, label = "Protein count", size = 3.2) +
geom_point(
data = size_legend,
aes(x = x, y = y),
size = size_legend$point_size,
color = "black"
) +
geom_text(
data = size_legend,
aes(x = x, y = y - 4.6, label = count),
size = 2.85
) +
scale_color_manual(values = c(cell_cols, setNames(cell_info$cell_state_color, cell_info$cell_state_color))) +
scale_fill_manual(values = cell_cols) +
coord_cartesian(xlim = c(0, 8.70), ylim = c(-9.5, 132), clip = "off") +
theme_void() +
theme(
legend.position = "none",
plot.margin = margin(8, 8, 8, 5)
)
ggsave("protein_cellstate_flow_bubble.png", p, width = 6.0, height = 8.2, dpi = 360, bg = "white")
ggsave("protein_cellstate_flow_bubble.pdf", p, width = 6.0, height = 8.2, bg = "white")完整代码
r
library(tidyverse)
library(scales)
links <- read_csv("protein_cellstate_links.csv", show_col_types = FALSE)
cell_info <- read_csv("cellstate_enrichment.csv", show_col_types = FALSE)
cell_levels <- cell_info$cell_state
links <- links |>
mutate(cell_state = factor(cell_state, levels = cell_levels))
cell_info <- cell_info |>
mutate(cell_state = factor(cell_state, levels = cell_levels))
cell_cols <- setNames(cell_info$cell_state_color, cell_info$cell_state)
panel_xmin <- 6.45
panel_xmax <- 8.35
panel_ymin <- 0
panel_ymax <- max(cell_info$cell_state_y + 4.8)
dot_df <- cell_info |>
mutate(
dot_x = rescale(
protein_ratio,
to = c(panel_xmin + 0.18, panel_xmax - 0.18),
from = c(0, 0.18)
),
dot_y = c(47, 35, 22, 11, 5),
dot_col = col_numeric(
palette = c("#1d2cff", "#7c169f", "#e23b2e", "#ff0000"),
domain = c(5, 32)
)(neg_log10_p),
dot_size = rescale(protein_count, to = c(2.4, 10.5), from = c(5, 65))
)
x_ticks <- tibble(
ratio = c(0.05, 0.10),
x = rescale(
ratio,
to = c(panel_xmin + 0.18, panel_xmax - 0.18),
from = c(0, 0.18)
)
)
color_legend <- tibble(
x = seq(panel_xmin + 0.18, panel_xmin + 0.85, length.out = 80),
y = 91,
value = seq(5, 32, length.out = 80)
) |>
mutate(
fill_col = col_numeric(
palette = c("#1d2cff", "#7c169f", "#e23b2e", "#ff0000"),
domain = c(5, 32)
)(value)
)
size_legend <- tibble(
x = c(panel_xmin + 0.18, panel_xmin + 0.70, panel_xmin + 1.26),
y = 76,
count = c(20, 40, 60),
point_size = c(3.7, 5.6, 7.6)
)
cell_bar_df <- cell_info |>
mutate(
xmin = 5.98,
xmax = 6.18,
ymin = cell_state_y - 4.8,
ymax = cell_state_y + 4.8
)
p <- ggplot() +
geom_curve(
data = links,
aes(
x = 0.64,
y = gene_y,
xend = 5.86,
yend = cell_state_y_link,
color = cell_state
),
curvature = -0.34,
linewidth = 0.24,
alpha = 0.36
) +
geom_segment(
data = links,
aes(x = 0.50, xend = 0.62, y = gene_y, yend = gene_y, color = tick_color),
linewidth = 1.0,
show.legend = FALSE
) +
geom_text(
data = links,
aes(x = 0.47, y = gene_y, label = protein_label),
hjust = 1,
size = 1.34,
color = "black"
) +
geom_rect(
data = cell_bar_df,
aes(xmin = xmin, xmax = xmax, ymin = ymin, ymax = ymax, fill = cell_state),
color = NA,
show.legend = FALSE
) +
geom_text(
data = cell_info,
aes(x = 5.83, y = cell_state_y, label = cell_state),
hjust = 1,
size = 4.0,
color = "black"
) +
annotate(
"rect",
xmin = panel_xmin,
xmax = panel_xmax,
ymin = panel_ymin,
ymax = panel_ymax,
fill = NA,
color = "black",
linewidth = 0.55
) +
geom_point(
data = dot_df,
aes(x = dot_x, y = dot_y),
color = dot_df$dot_col,
size = dot_df$dot_size
) +
geom_segment(
data = x_ticks,
aes(x = x, xend = x, y = panel_ymin, yend = panel_ymin - 1.15),
linewidth = 0.45,
color = "black"
) +
geom_text(
data = x_ticks,
aes(x = x, y = panel_ymin - 3.0, label = sprintf("%.2f", ratio)),
size = 2.85,
color = "black"
) +
annotate(
"text",
x = mean(c(panel_xmin, panel_xmax)),
y = panel_ymin - 7.0,
label = "Protein ratio",
size = 3.8
) +
geom_tile(
data = color_legend,
aes(x = x, y = y),
fill = color_legend$fill_col,
width = 0.012,
height = 1.6
) +
annotate("text", x = panel_xmin + 0.54, y = 95.0, label = "-log10(p-value)", size = 3.4) +
annotate("text", x = panel_xmin + 0.25, y = 87.7, label = "10", size = 3.0) +
annotate("text", x = panel_xmin + 0.53, y = 87.7, label = "20", size = 3.0) +
annotate("text", x = panel_xmin + 0.78, y = 87.7, label = "30", size = 3.0) +
annotate("text", x = panel_xmin + 0.62, y = 81.5, label = "Protein count", size = 3.2) +
geom_point(
data = size_legend,
aes(x = x, y = y),
size = size_legend$point_size,
color = "black"
) +
geom_text(
data = size_legend,
aes(x = x, y = y - 4.6, label = count),
size = 2.85
) +
scale_color_manual(values = c(cell_cols, setNames(cell_info$cell_state_color, cell_info$cell_state_color))) +
scale_fill_manual(values = cell_cols) +
coord_cartesian(xlim = c(0, 8.70), ylim = c(-9.5, 132), clip = "off") +
theme_void() +
theme(
legend.position = "none",
plot.margin = margin(8, 8, 8, 5)
)
ggsave("protein_cellstate_flow_bubble.png", p, width = 6.0, height = 8.2, dpi = 360, bg = "white")
ggsave("protein_cellstate_flow_bubble.pdf", p, width = 6.0, height = 8.2, bg = "white")复现结果