vasp如何计算金属的铁电极化强度?
这个问题本身就是一个非常好的"概念陷阱"。
严格来说,大多数金属不存在传统意义上的铁电极化,因此 VASP 也无法直接计算金属的铁电极化强度。
真正需要回答的问题其实是:
"为什么金属不能定义铁电极化?如果实验上报道了金属铁电,又应该如何计算?"
为什么绝缘体能定义极化?
现代极化理论(Modern Theory of Polarization)认为:
铁电极化不是
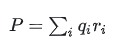
这种经典偶极矩。
而是
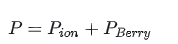
其中电子部分来自 Berry Phase:
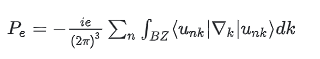
这里要求:
所有占据能带与空带之间存在带隙。
为什么?
因为 Berry phase 必须定义在
完整占据的电子子空间(occupied manifold)
上。二、为什么金属不能?
金属最大的特点:
费米能级穿过能带。
于是
occupied band
和
unoccupied band
不断交换。
Berry connection
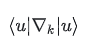
已经不能定义全局连续规范。
因此:
金属不存在严格定义的 Berry Phase 极化。
也就是说
VASP 的
LCALCPOL = .TRUE.对于普通金属根本没有理论基础。
通常程序都会直接报错或者得到没有物理意义的数据。
为什么有人说"金属铁电"?
这里就进入近年来比较热门的话题。
例如
-
LiOsO3
-
WTe2
-
MoTe2
-
某些二维 Janus 金属
-
某些极性金属
这些论文都会写
Ferroelectric metal
其实要分清:
它们具有的是
极性晶体结构(polar distortion)
而不是
可严格定义 Berry 极化。
因为自由电子会屏蔽静电场。
因此:
真正可以讨论的是
-
极性位移
-
非中心对称结构
-
开关能垒
-
声子软模
-
极化路径
而不是 Berry Phase 数值。
所以很多论文都会避免写
polarization = xx μC/cm²
而写
polar displacement
或者
polar distortion amplitude
如果一定要计算"金属铁电极化",怎么办?
实际上有几种不同的方法。
人为打开带隙
例如:
DFT+U
或者
HSE
使体系暂时变成绝缘体。
然后计算
LCALCPOL = .TRUE.得到 Berry Phase。
很多论文都是这么干。
但是一定要说明:
这是参考极化(reference polarization),不是金属真实极化。
Born Effective Charge
计算
LEPSILON = .TRUE.得到
Born effective charge
然后
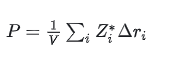
这个方法很多极性金属都会使用。
因为
Born charge
描述的是
极性位移产生的偶极。
不过严格来说:
金属中 Born charge 本身也不再严格定义。
很多时候只是近似。
点电荷模型
利用
Bader Charge
或者
DFT 电荷
近似
P=\\frac1V\\sum_i q_i\\Delta r_i
这个方法误差较大。
通常只讨论趋势。
构造绝缘参考相(推荐)
这是现在很多高水平论文喜欢采用的方法。
例如:
把
极性结构
和
中心对称结构
之间建立连续路径。
计算
-
位移模式
-
能量曲线
-
极化路径
如果中间经过绝缘态,
即可利用 Berry Phase 得到参考极化。
因此文献常写:
polarization estimated from insulating reference phase
而不是
metal polarization。
VASP 能做哪些计算?
对于普通金属:
不能直接使用
LCALCPOL = .TRUE.因为 Berry Phase 理论不适用。
更推荐计算:
-
极性位移(Polar displacement)
-
声子谱(PHONOPY)
-
双稳态势能面(NEB 或冻结声子)
-
Born Effective Charge(若体系允许)
-
对称性分析(ISODISTORT、AMPLIMODES)
-
Wannier 中心变化(特殊情况下)
这些共同证明体系具有极性或铁电相关行为。
很多初学者误以为 VASP 的
LCALCPOL是一个"极化计算按钮",实际上它只是现代极化理论(Berry Phase)的实现,而现代极化理论建立在绝缘体的完整占据电子子空间之上。因此,"如何计算金属的铁电极化强度"这个问题本身就需要先修正为:"如何表征金属中的极性畸变或铁电行为"。只有把理论对象弄清楚,后续的计算方法才有物理意义。