数据和代码获取:请查看主页个人信息!!!
原核生物分类单元功能注释 Functional Annotation of Prokaryotic Taxa ( FAPROTAX **)**是美国俄勒冈大学生物学系Louca实验室开发的一个数据库,该数据库利用当前有关培养菌株的文献,将原核分支(如属或种)映射到已建立的代谢或其他生态相关功能。
FAPROTAX概要:
-
80多种功能,如硝酸盐呼吸、产甲烷、发酵或植物致病。
-
7600多个功能注释,涵盖4600多个分类群。
-
利用文献包括贝格雷的系统细菌学手册、原核生物和国际系统细菌学杂志。
FAPROTAX的潜在应用:
-
16S标记基因数据的生态学解释。
-
用于功能社区分析的环境鸟枪测序(宏基因组学)的廉价替代品。
-
补充宏基因组学,例如解决社区基因内容解释中的歧义。
-
快速识别具有特定代谢表型(例如所有已知或假定的产甲烷菌)的样本中的分类群。

FAPROTAX包括一个软件,用于根据样本中确定的分类群,将分类微生物群落特征(例如,以OTU表的形式)转换为假定的功能特征。Louca实验室基于python开发了配套软件,官方软件地址:http://www.loucalab.com/
今天我们使用R语言实现FAPROTAX原核生物分类单元的功能注释分析,接下来我们来进行分析和可视化展示:

Step1:数据载入
rm(list=ls())pacman::p_load(tidyverse,microeco,aplot,ggsci)
feature_table <- read.csv('feature_table.csv', row.names = 1)sample_table <- read.csv('sample_table.csv', row.names = 1)tax_table <- read.csv('tax_table.csv', row.names = 1)Step2:构建 microtable 对象
# 创建microtable对象dataset <- microtable$new(sample_table = sample_table, otu_table = feature_table, tax_table = tax_table)
这里的操作与前面介绍的几期内容一致:R语言实战:microeco包轻松实现微生物组LEfSe分析;随机森林分析:R包microeco实现微生物组随机森林分析及重要变量的选择
Step3:执行FAPROTAX注释分析
t2 <- trans_func$new(dataset)t2$cal_spe_func(prok_database = "FAPROTAX")t2$res_spe_func[1:5, 1:2]
# 具有相同特征的OTU的百分比可以反映该功能在群落中的功能冗余程度。t2$cal_spe_func_perc(abundance_weighted = FALSE)t2$res_spe_func_perc[1:5, 1:2]
原作者在文章**Decoupling function and taxonomy in the global ocean microbiome(DOI: 10.1126/science.aaf4507)**中使用气泡图对注释结果进行可视化:
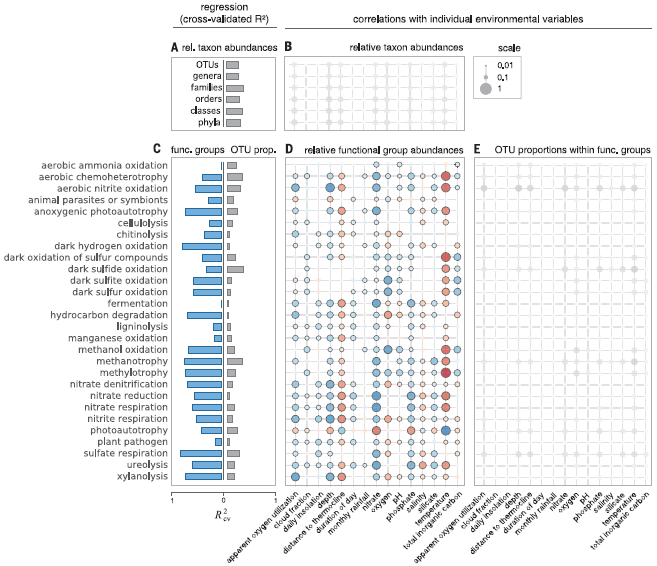
我们这里同样使用气泡图展示注释分析结果:
Step4:气泡图展示注释结果
t2$res_spe_func_perc %>% select(1:30) %>% scale() %>% as.data.frame() %>% rownames_to_column('sample') %>% melt(id.vars = 'sample') %>% ggplot(aes(sample, variable)) + labs(x="",y="") + geom_point(aes(size=abs(value),color=value)) + scale_size_area(max_size = 2) + scale_color_gsea() + theme_classic(base_size = 4.5) + theme(axis.text.x=element_text(angle=45,vjust=1, hjust=1))ggsave('pic_bo_cor.png', width = 7, height = 4)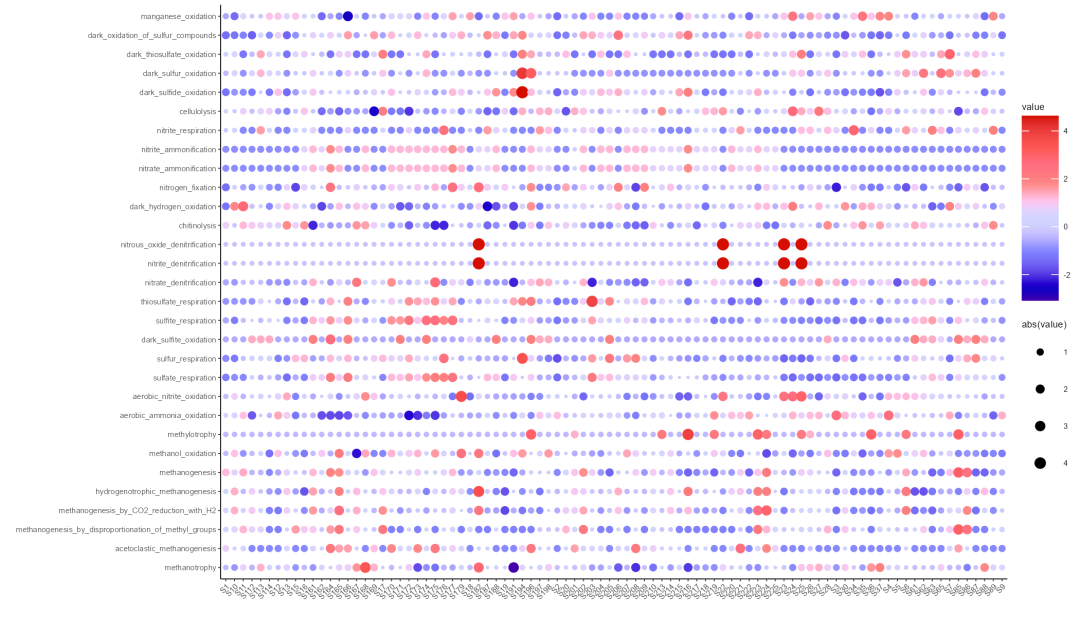
当然,也可以参考microeco包作者推荐的分析和可视化方案,将微生物模块化分组,结合功能注释分析,有助于找到核心的特征微生物。
Step5:展示网络模块中每个特征的OTU百分比
network <- trans_network$new(dataset = dataset, cal_cor = "base", taxa_level = "OTU", filter_thres = 0.0001, cor_method = "spearman")network$cal_network(p_thres = 0.01, COR_cut = 0.7)network$cal_module()# convert module info to microtable objectmeco_module <- network$trans_comm(use_col = "module")meco_module_func <- trans_func$new(meco_module)meco_module_func$cal_spe_func(prok_database = "FAPROTAX")meco_module_func$cal_spe_func_perc(abundance_weighted = FALSE)meco_module_func$plot_spe_func_perc(order_x = paste0("M", 1:10))
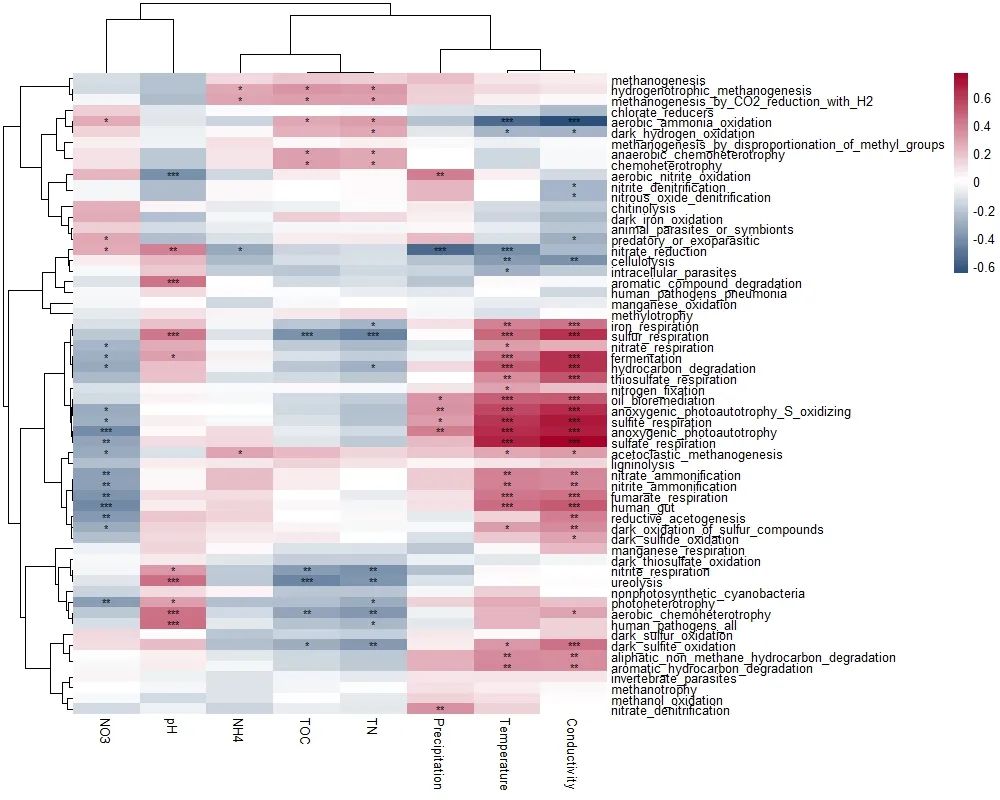
Step6:群落功能与环境因子相关性
t3 <- trans_env$new(dataset = dataset, add_data = env_data_16S[, 4:11])
t3$cal_cor(add_abund_table = t2$res_spe_func_perc, cor_method = "spearman")
t3$plot_cor(pheatmap = T)