微阵列和RNA-Seq等基因表达研究常常会产生成百上千个差异表达基因 (DEG)。要了解这种海量数据集的生物学意义变得非常困难,尤其是在存在多种条件和比较的情况下。
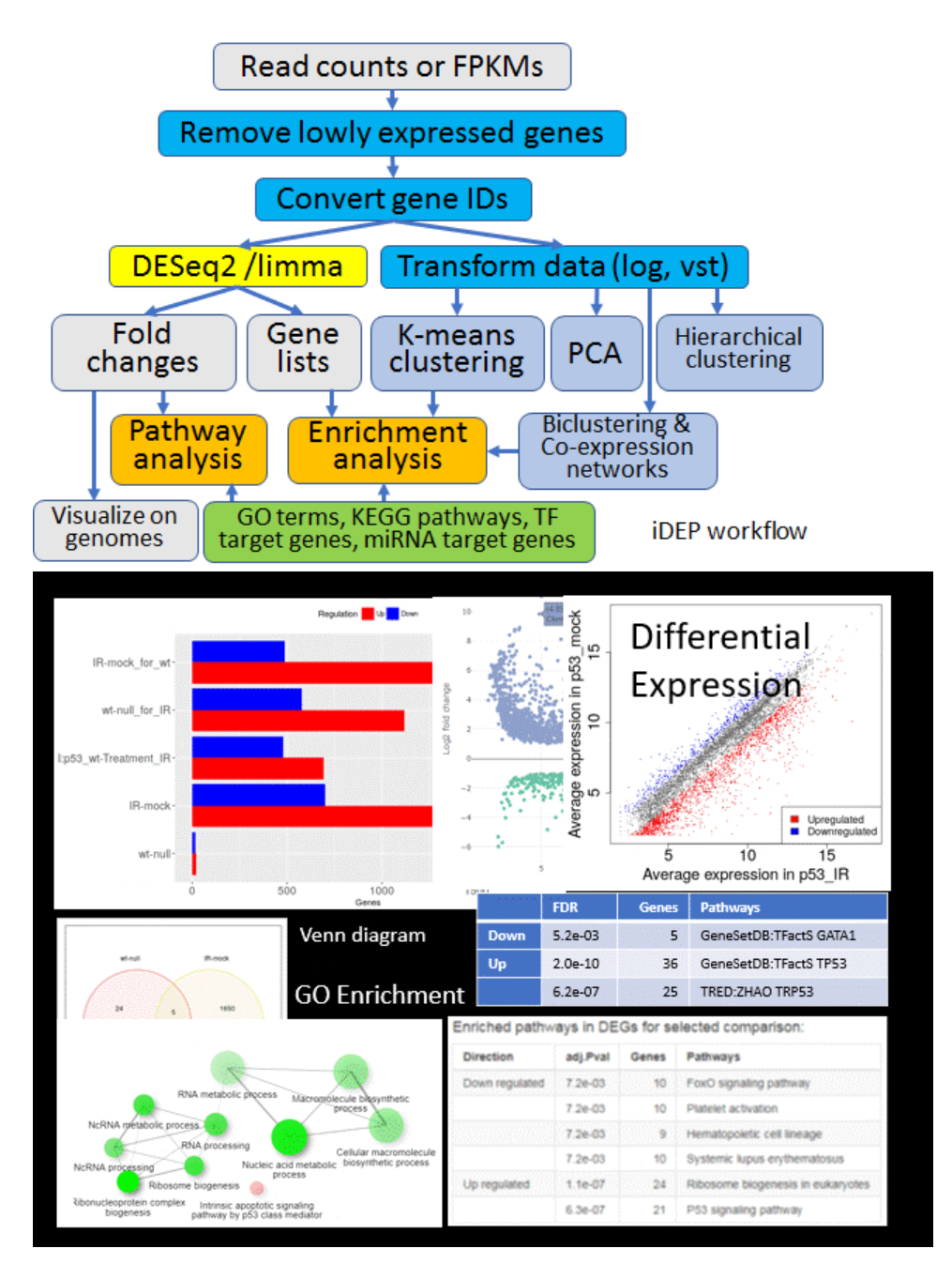
▲ 常规的RNA测序分析流程。图片来源:10.1186/s12859-018-2486-6。
为此,来自英属哥伦比亚大学的研究者开发了一款软件包pathlinkR ,于2024年9月16号发表于PLoS Comput Biol****IF: 3.8。该软件包集成了通路富集和蛋白-蛋白相互作用网络,基于差异基因表达结果的可视化和下游分析,可以帮助研究人员从基因表达研究中揭示潜在的生物学和病理生理学。

▲ DOI: 10.1371/journal.pcbi.1012422
本文简要介绍一下pathlinkR的示例运行,想进一步了解的铁子可以阅读以下链接:
R包安装与示例数据
该软件包中提供了一个基因差异表达结果的示例数据集,即对象exampleDESeqResults。这是一个由2个数据框组成的列表,是使用软件包DESeq2中的results()函数生成的。数据来自一项RNA-Seq研究,该研究调查了COVID-19和非 COVID-19 败血症患者在入院时(T1)与在重症监护室约一周后(T2)的情况,并进行了时间比较(即T2 vs T1)。
可以使用以下代码安装引用R包,并加载示例数据集:
python
# We'll also be using some functions from dplyr
# BiocManager::install("pathlinkR")
library(dplyr)
library(pathlinkR)
## A quick look at the DESeq2 results table
data("exampleDESeqResults")
knitr::kable(head(exampleDESeqResults[[1]]))
# baseMean log2FoldChange lfcSE stat pvalue padj
#ENSG00000000938 16292.64814 -0.5624954 0.1458274 -3.857268 0.0001147 0.0013531
#ENSG00000002586 1719.51750 0.4501181 0.1520122 2.961066 0.0030658 0.0153820
#ENSG00000002919 870.64168 -0.2445729 0.1249293 -1.957690 0.0502664 0.1236844
#ENSG00000002933 266.65476 0.8838310 0.2093628 4.221528 0.0000243 0.0004313
#ENSG00000003249 11.43282 1.3287128 0.2881385 4.611369 0.0000040 0.0001200
#ENSG00000003509 207.88545 -0.1825614 0.1763556 -1.035189 0.3005807 0.4453252此外,该包还包含了一个使用通路富集软件包Sigora,基于Reactome数据库的富集结果注释。有关Reactome通路和Sigora软件包的更多信息,可以参阅等会的通路富集部分。
python
data("sigoraDatabase")
head(sigoraDatabase)
# A tibble: 6 × 4
# pathwayId pathwayName ensemblGeneId hgncSymbol
# <chr> <chr> <chr> <chr>
#1 R-HSA-114608 "Platelet degranulation " ENSG00000121410 A1BG
#2 R-HSA-6798695 "Neutrophil degranulation" ENSG00000121410 A1BG
#3 R-HSA-76002 "Platelet activation, signaling and aggregation" ENSG00000121410 A1BG
#4 R-HSA-76005 "Response to elevated platelet cytosolic Ca2+" ENSG00000121410 A1BG
#5 R-HSA-156580 "Phase II - Conjugation of compounds" ENSG00000156006 NAT2
#6 R-HSA-211859 "Biological oxidations" ENSG00000156006 NAT2 pathlinkR使用
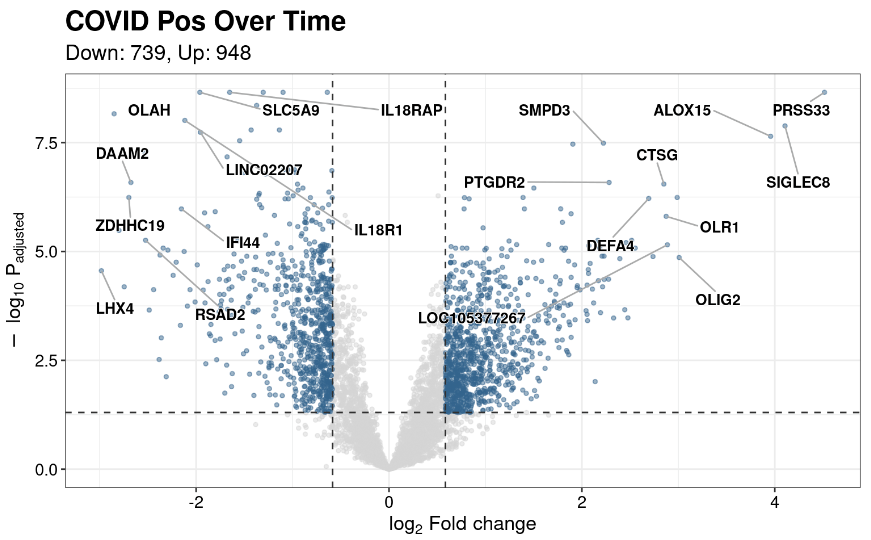
① 火山图:
python
eruption(
rnaseqResult=exampleDESeqResults[[1]],
title=names(exampleDESeqResults[1])
)
eruption函数支持多种参数。比如,我们也可以指定高亮某些基因:
python
data("sigoraDatabase")
interferonGenes <- sigoraDatabase %>%
filter(pathwayName == "Interferon Signaling") %>%
pull(ensemblGeneId)
eruption(
rnaseqResult=exampleDESeqResults[[1]],
title=names(exampleDESeqResults[1]),
highlightGenes=interferonGenes,
highlightName="Interferon genes (red)",
label="highlight",
nonlog2=TRUE,
n=10
)上方参数达成的效果是:
- 突出干扰素基因并简要说明
- 将x轴从log2FC转换为非log2FC
- 仅标出属于干扰素通路的前 10 个上调和下调基因
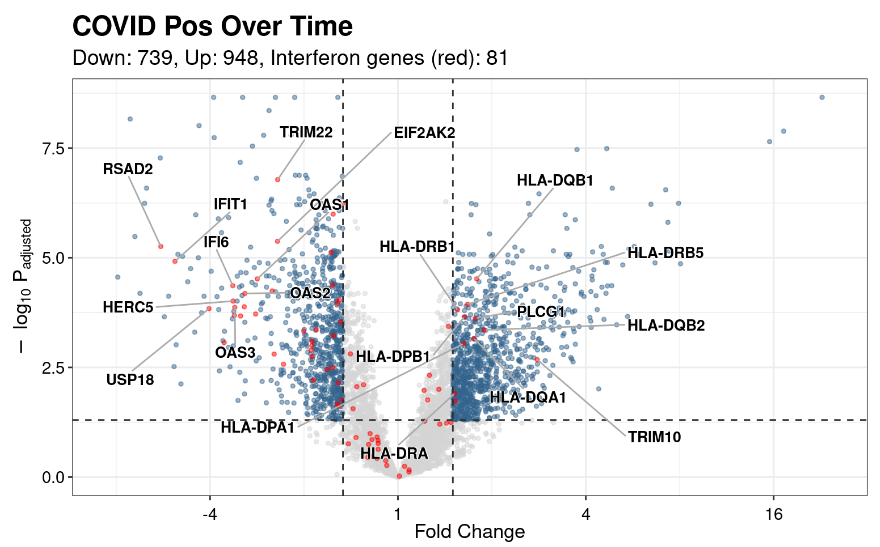
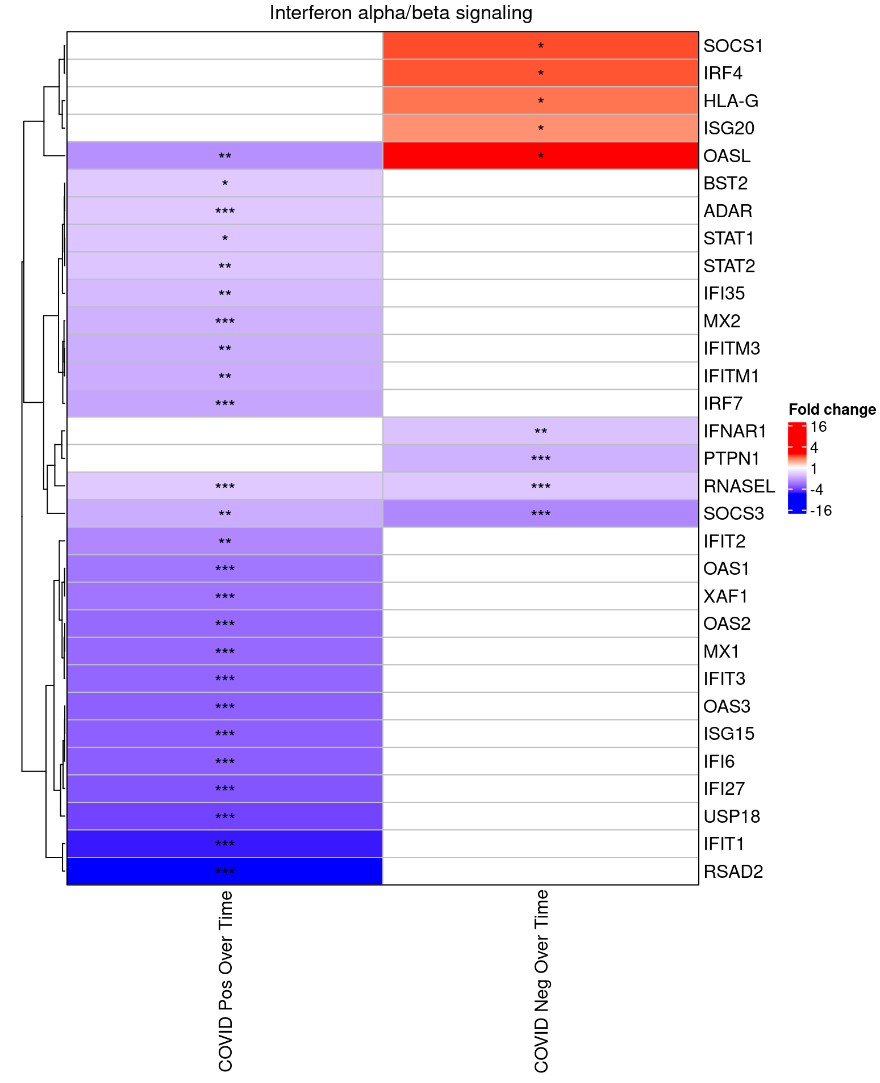
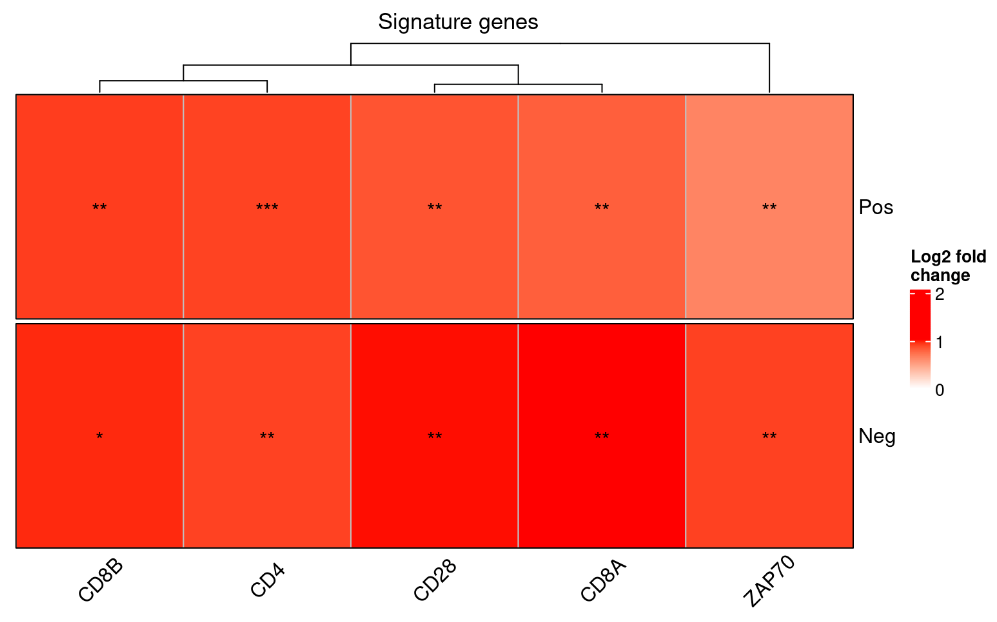
② 热图:
除了创建火山图外,可以使用参与特定通路(例如通过pathwayEnrichment()函数确定为重要的通路)的基因热图来可视化 DEGs。函数plotFoldChange()通过接收DESeq2::results()的输入列表并创建组成基因的FC (Fold Chage)热图来实现这一目的。
python
plotFoldChange(
exampleDESeqResults,
pathName="Interferon alpha/beta signaling"
)
另一个例子使用了更多参数,定制化程度更高:
- 缩短每次比较的名称
- 提供可视化基因的自定义列表,并添加内容丰富的标题
- 在热图单元中使用log2FC
- 交换默认的行和列,并在行之间添加分割(比较)
python
exampleDESeqResultsRenamed <- setNames(
exampleDESeqResults,
c("Pos", "Neg")
)
plotFoldChange(
exampleDESeqResultsRenamed,
manualTitle="Signature genes",
genesToPlot=c("CD4", "CD8A","CD8B", "CD28", "ZAP70"),
geneFormat="hgnc",
colSplit=c("Pos", "Neg"),
log2FoldChange=TRUE,
colAngle=45,
clusterRows=FALSE,
clusterColumns=TRUE,
invert=TRUE
)
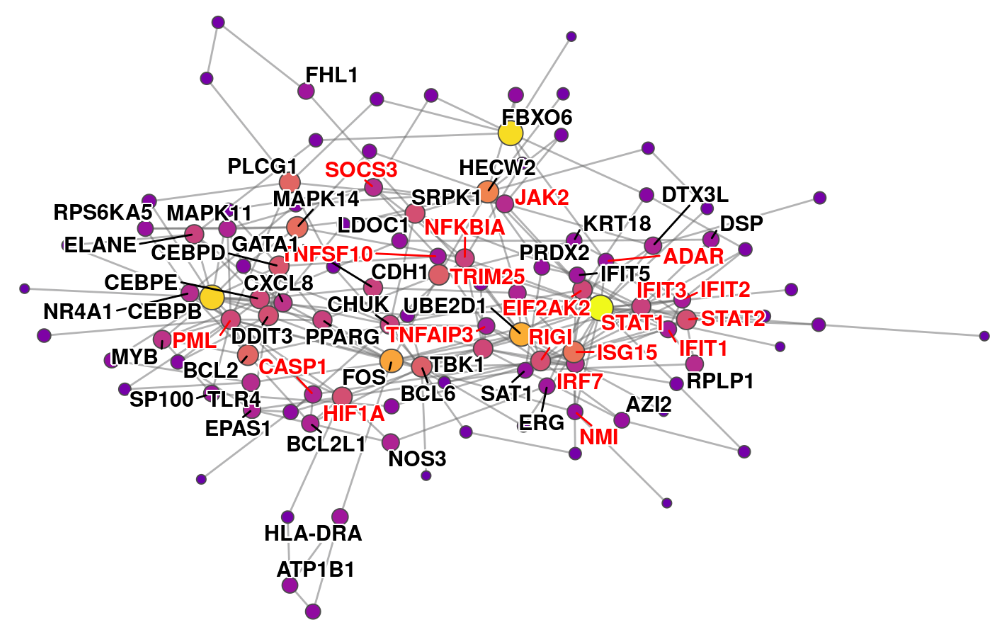
③ 构建和可视化PPI网络:
pathlinkR包含用于构建和可视化蛋白质-蛋白质相互作用(PPI)网络的工具。**在这里,利用从InnateDB收集到的PPI数据,生成基因表达分析中发现的DE基因之间的相互作用列表。**可以继续观察COVID阳性患者的DEGs随时间变化的情况,利用显著的DEGs构建PPI网络。由于输入的数据包括所有测量基因,因此需要使用filterInput=TRUE选项来确保只使用通过标准阈值的基因来构建网络。此外,还可以通过指定fillType="foldChange"来为节点着色,以指示其失调的方向(即上调或下调)。
python
exNetwork <- ppiBuildNetwork(
rnaseqResult=exampleDESeqResults[[1]],
filterInput=TRUE,
order="zero"
)
ppiPlotNetwork(
network=exNetwork,
title=names(exampleDESeqResults)[1],
fillColumn=LogFoldChange,
fillType="foldChange",
label=TRUE,
labelColumn=hgncSymbol,
legend=TRUE
)
带有蓝色标签的节点(如 STAT1、FBXO6、CDH1 等)是网络中的枢纽,即具有较高BC分值的基因。用于确定中心节点的统计量可在ppiBuildNetwork()中使用hubMeasure选项进行设置。
随后还对这个标准网络进行一些调整。ppiBuildNetwork()的输出是一个tidygraph对象,这意味着可以使用一套dplyr函数来进行调整。比如,添加一列来表示DEG是否来自干扰素通路(如上定义),并只突出显示这些基因:
python
exNetworkInterferon <- mutate(
exNetwork,
isInterferon = if_else(name %in% interferonGenes, "y", "n")
)
ppiPlotNetwork(
network=exNetworkInterferon,
title=names(exampleDESeqResults)[1],
fillColumn=isInterferon,
fillType="categorical",
catFillColours=c("y"="red", "n"="grey"),
label=TRUE,
labelColumn=hgncSymbol,
legend=TRUE,
legendTitle="Interferon\ngenes"
)
④ ppi的网络富集:
pathlinkR包含两个函数用于进一步分析PPI网络。首先,ppiEnrichNetwork()将使用网络中的节点富集的Reactome通路或Hallmark基因集(有关于富集的详细内容可以参照下一节):
python
exNetworkPathways <- ppiEnrichNetwork(
network=exNetwork,
analysis="hallmark",
filterResults="default",
geneUniverse = rownames(exampleDESeqResults[[1]])
)
exNetworkPathways
# A tibble: 5 × 10
# pathwayId pathwayName pValue pValueAdjusted genes numCandidateGenes
# <chr> <chr> <dbl> <dbl> <chr> <dbl>
#1 INTERFERON GAMMA R... INTERFERON... 1.68e-7 0.00000791 HLA-... 45
#2 INTERFERON ALPHA R... INTERFERON... 2.20e-6 0.0000518 IRF7... 30
#3 TNFA SIGNALING VIA... TNFA SIGNA... 3.58e-5 0.000560 CEBP... 32
#4 APOPTOSIS APOPTOSIS 1.01e-3 0.0118 CASP... 21
#5 INFLAMMATORY RESPO... INFLAMMATO... 1.98e-3 0.0186 MXD1... 28
# ℹ 4 more variables: numBgGenes <dbl>, geneRatio <dbl>, totalGenes <int>,
# topLevelPathway <chr>随后,函数ppiExtractSubnetwork()可以从起始网络中提取最小连接的子网络,并将富集通路中的基因作为提取的 "起始 "节点。例如,下面使用上述Hallmark富集的结果,从 "干扰素γ反应 "项中提取出一个由基因组成的子网络,然后绘制这个缩小的网络,同时突出显示通路中的基因:
exSubnetwork <- ppiExtractSubnetwork(
network=exNetwork,
pathwayEnrichmentResult=exNetworkPathways,
pathwayToExtract="INTERFERON GAMMA RESPONSE"
)
ppiPlotNetwork(
network=exSubnetwork,
fillType="oneSided",
fillColumn=degree,
label=TRUE,
labelColumn=hgncSymbol,
legendTitle = "Degree"
)
⑤ 进行通路富集:
路径富集分析的核心是过度代表性分析(over-representation),即在我们关注的差异表达基因(DEG)列表中,属于特定通路的基因是否比预期出现得更多。简单来说,这种分析通过比较通路中DEG的比例和数据库中所有基因的比例来实现。pathlinkR主要使用 Reactome 数据库来完成这一分析。**然而,这种方法假设每个基因在每条通路中的作用是"均等"的,忽视了基因可能在多个通路中发挥不同作用的问题。**这会导致结果中出现大量相似的通路,或与生物学不相关的"假阳性"通路,增加了解读结果的难度。
为了解决这一问题,pathlinkR使用了Sigora提出的一种基于唯一基因对的方法,可以减少因多功能基因而导致的通路重叠,聚焦于与生物学现象更相关的通路。pathlinkR的pathwayEnrichment()函数默认采用这种方法,并可选择fgsea包实现的基因集富集分析,同时支持Reactome和Hallmark数据集。pathwayEnrichment()函数以多个DESeq2::results()数据框作为输入,默认按上调和下调基因进行富集分析。对于analysis="sigora"的选项,还需要提供基因对签名库(GPS repository),默认使用来自Sigora的Reactome库:
python
enrichedResultsSigora <- pathwayEnrichment(
inputList=exampleDESeqResults,
analysis="sigora",
filterInput=TRUE,
gpsRepo="default"
)
head(enrichedResultsSigora)
# A tibble: 6 × 12
# comparison direction pathwayId pathwayName pValue pValueAdjusted genes
# <chr> <chr> <chr> <fct> <dbl> <dbl> <chr>
#1 COVID Pos Over ... Up R-HSA-38... Chemokine ... 7.04e-46 7.05e-43 CCR3...
#2 COVID Pos Over ... Up R-HSA-19... Immunoregu... 5.46e-45 5.46e-42 CD1A...
#3 COVID Pos Over ... Up R-HSA-38... Costimulat... 2.56e-40 2.56e-37 CD28...
#4 COVID Pos Over ... Up R-HSA-67... Neutrophil... 7.88e-31 7.89e-28 ABCA...
#5 COVID Pos Over ... Up R-HSA-20... Cell surfa... 3.55e-21 3.55e-18 ATP1...
#6 COVID Pos Over ... Up R-HSA-14... Alpha-defe... 1.02e-20 1.02e-17 CD4;...
# ℹ 5 more variables: numCandidateGenes <dbl>, numBgGenes <int>,
# geneRatio <dbl>, totalGenes <int>, topLevelPathway <chr>注意上面的direction是分开的,也就是上调基因、下调基因单独富集。如果想同时测试上调基因和下调基因,只需设置split=FALSE参数。
对于那些仍然偏好传统过度表现分析的人,作者也提供了参数analysis="reactomepa",使用基于余光创老师开发的ReactomePA包进行富集:
python
data("mappingFile")
exGeneUniverse <- mappingFile %>%
filter(ensemblGeneId %in% rownames(exampleDESeqResults[[1]])) %>%
pull(entrezGeneId)
enrichedResultsRpa <- pathwayEnrichment(
inputList=exampleDESeqResults,
analysis="reactomepa",
filterInput=TRUE,
split=TRUE,
geneUniverse=exGeneUniverse
)
head(enrichedResultsRpa)
# A tibble: 6 × 12
# comparison direction pathwayId pathwayName pValue pValueAdjusted genes
# <chr> <chr> <chr> <chr> <dbl> <dbl> <chr>
#1 COVID Pos Over ... Up R-HSA-20... Generation... 3.63e-10 0.000000219 PLCG...
#2 COVID Pos Over ... Up R-HSA-20... Translocat... 1.51e- 9 0.000000302 HLA-...
#3 COVID Pos Over ... Up R-HSA-38... PD-1 signa... 1.51e- 9 0.000000302 HLA-...
#4 COVID Pos Over ... Up R-HSA-20... Phosphoryl... 1.22e- 7 0.0000184 HLA-...
#5 COVID Pos Over ... Up R-HSA-38... Costimulat... 1.22e- 6 0.000146 CD28...
#6 COVID Pos Over ... Up R-HSA-20... TCR signal... 3.61e- 6 0.000321 PLCG...
# ℹ 5 more variables: numCandidateGenes <dbl>, numBgGenes <dbl>,
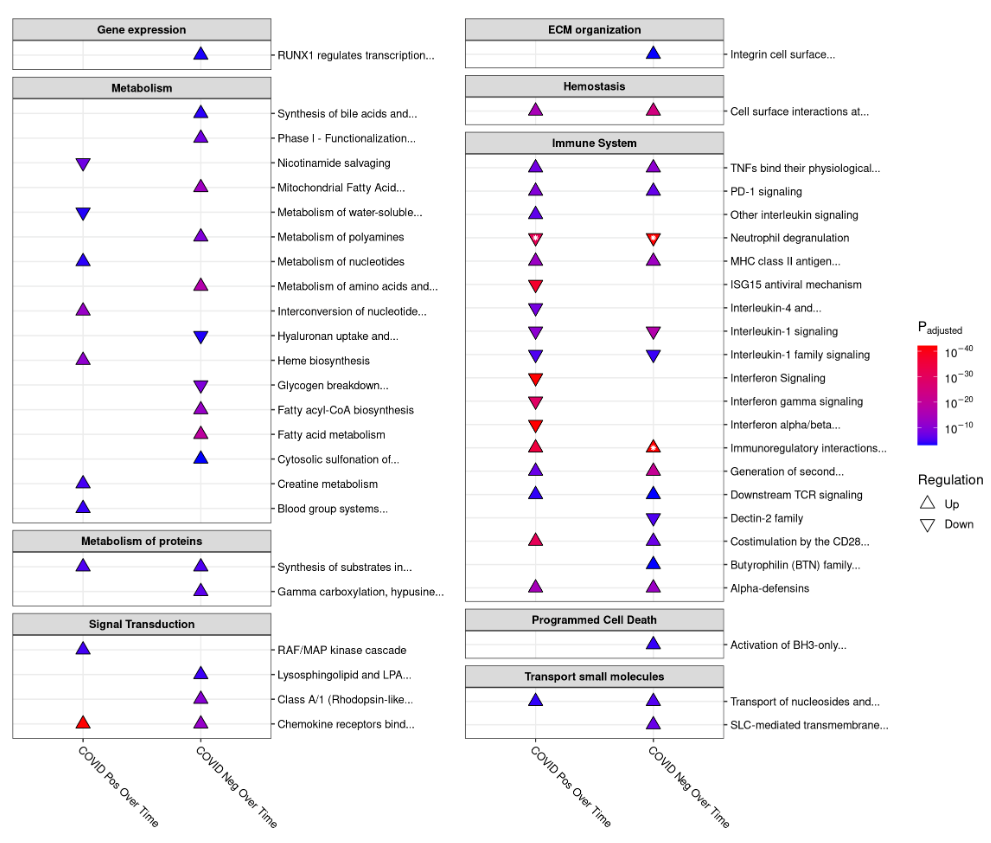
# geneRatio <dbl>, totalGenes <int>, topLevelPathway <chr>富集结果可以通过以下代码进行可视化(想了解更多参数可以看看官网):
python
pathwayPlots(
pathwayEnrichmentResults=enrichedResultsSigora,
columns=2
)
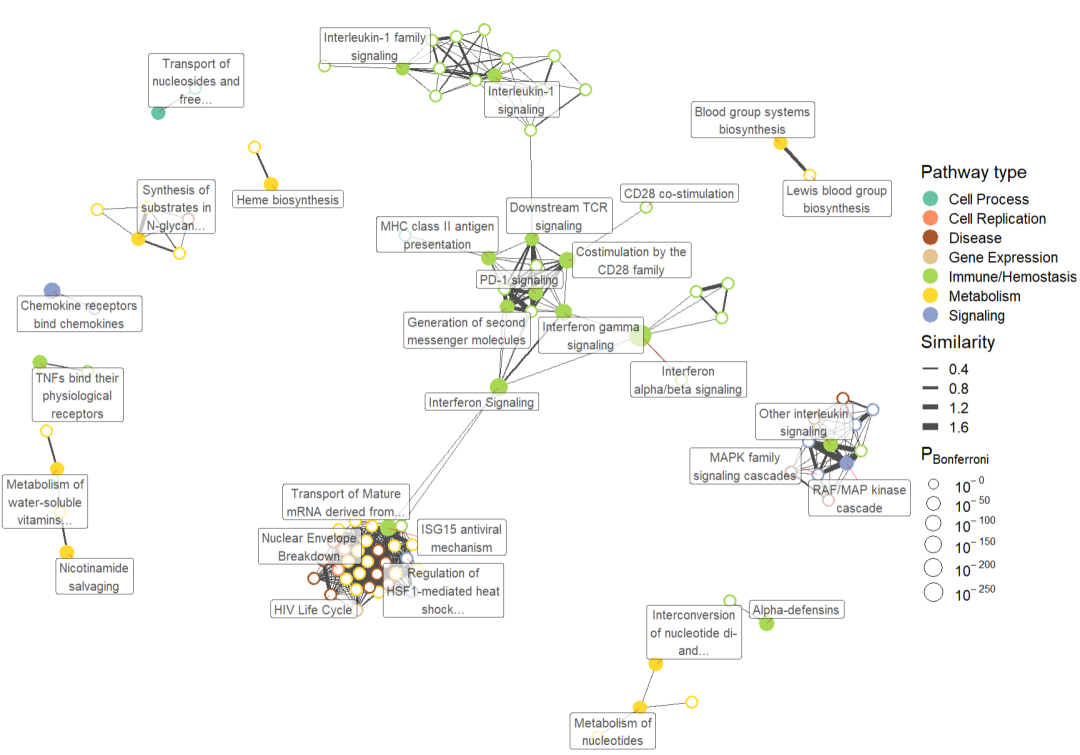
⑥ 根据富集结果生成网络
pathlinkR包括将基于Reactome 方法("sigora "或 "reactomepa")的通路富集结果转化为网络的功能,利用分配给每个通路的基因的重叠性来确定它们之间的相似性。在这些网络中,每条通路都是一个节点,它们之间的连接或边缘是通过距离度量确定的。可以设置一个阈值,将具有最小相似度的两条途径视为相连,并在它们的节点之间画一条边。默认情况下,该包提供了一个预先计算的Reactome通路距离矩阵,该矩阵是使用 Jaccard 距离生成的,但也支持使用多种距离度量 。一旦创建了通路相互作用的 "基础",就可以使用createPathnet函数构建通路网络:
python
data("sigoraExamples") # 加载富集结果
pathwayDistancesJaccard <- getPathwayDistances(pathwayData = sigoraDatabase)
startingPathways <- pathnetFoundation(
mat=pathwayDistancesJaccard,
maxDistance=0.8
)
# Get the enriched pathways from the "COVID Pos Over Time" comparison
exPathwayNetworkInput <- sigoraExamples %>%
filter(comparison == "COVID Pos Over Time")
myPathwayNetwork <- pathnetCreate(
pathwayEnrichmentResult=exPathwayNetworkInput,
foundation=startingPathways
)随后可以使用两种方案可视化,第一种是静态网络:
pathnetGGraph(
myPathwayNetwork,
labelProp=0.1,
nodeLabelSize=4,
nodeLabelOverlaps=8,
segColour="red"
)
第二种是交互式网络:
python
pathnetVisNetwork(myPathwayNetwork)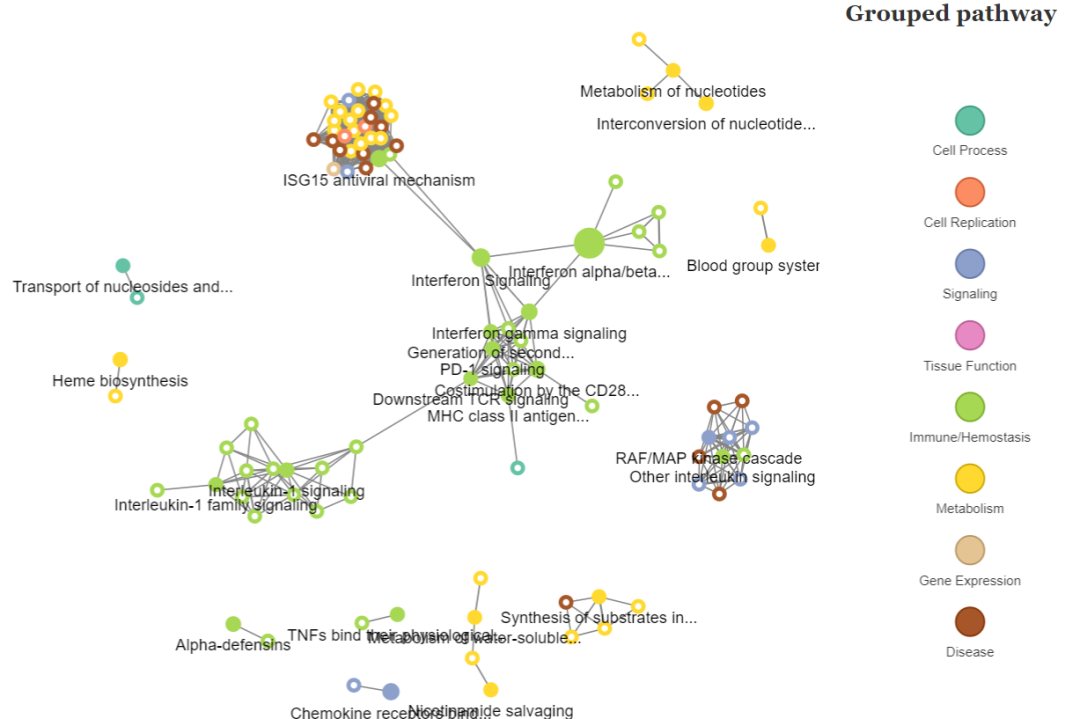
最后,可以使用以下代码在生成通路网络信息时只使用 DEGs:
python
candidateData <- sigoraExamples %>%
filter(comparison == "COVID Pos Over Time") %>%
select(pathwayId, genes) %>%
tidyr::separate_rows(genes, sep=";") %>%
left_join(
sigoraDatabase,
by=c("pathwayId", "genes" = "hgncSymbol"),
multiple="all"
) %>%
relocate(pathwayId, ensemblGeneId, "hgncSymbol"=genes, pathwayName) %>%
distinct()
## Now that we have a smaller table in the same format as sigoraDatabase, we
## can construct our own matrix of pathway distances
candidateDistData <- getPathwayDistances(
pathwayData=candidateData,
distMethod="jaccard"
)
candidateStartingPathways <- pathnetFoundation(
mat=candidateDistData,
maxDistance=0.9
)
candidatesAsNetwork <- pathnetCreate(
pathwayEnrichmentResult=filter(
sigoraExamples,
comparison == "COVID Pos Over Time"
),
foundation=candidateStartingPathways,
trim=FALSE
)
#
pathnetGGraph(
myPathwayNetwork,
labelProp=0.1,
nodeLabelSize=4,
nodeLabelOverlaps=8,
segColour="red"
)