文献介绍

文献题目: 使用 STAMP 对空间转录组进行可解释的空间感知降维
研究团队: 陈金妙(新加坡科学技术研究局)
发表时间: 2024-10-15
发表期刊: Nature Methods
影响因子: 36.1(2024年)
DOI: 10.1038/s41592-024-02463-8
摘要
空间转录组学产生具有空间背景的高维基因表达测量。获得此类数据的具有生物学意义的低维表示对于有效解释和下游分析至关重要。在这里,作者提出了主题建模揭示空间模式的空间转录组学分析(STAMP),这是一种基于深度生成模型的可解释的空间感知降维方法,可返回生物相关的低维空间主题和相关基因模块。STAMP 可以分析从单个部分到多个部分、从不同技术到时间序列数据的数据,返回与已知生物域和相关基因模块匹配的主题,其中包含已建立的高排名标记。在肺癌样本中,STAMP 以比原始注释更高的分辨率用支持标记描绘了细胞状态,并发现了集中在肿瘤边缘外部的癌症相关成纤维细胞。在小鼠胚胎发育的时间序列数据中,STAMP 解开了肝脏内红骨髓造血和肝细胞的发育轨迹。STAMP 具有高度可扩展性,可以处理超过 500,000 个细胞。
前言
空间转录组学是探索组织结构的一项重要实验技术,因为它可以捕获基因表达谱,同时保留其空间背景。因此,需要空间感知分析来整合基因表达和空间信息,以充分利用这些数据。在常用的工作流程中,降维形成初始处理步骤,这一步骤尤其重要,因为其捕获相关数据变异性的准确性会影响数据可视化和下游分析,例如用于识别生物学相关空间域或细胞类型的聚类。此后,通常进行差异表达分析和细胞注释。或者,可解释的降维是一个有吸引力的选择,它利用每个基因对低维嵌入的计算贡献。通过在空间坐标上可视化这种嵌入并检查贡献的输入基因,人们可以破译解剖区域或细胞类型,而无需聚类和差异表达分析。与黑盒建模方法相比,这也使人们对降维的生物学相关性更有信心。
单细胞分析中经常使用主成分分析 (PCA)、非负矩阵分解 (NMF) 和潜在狄利克雷分配 (LDA) 等经典降维方法。它们返回可解释的嵌入,但可能缺乏表现力并且不包含空间信息。或者,自动编码器 (AE) 方法,例如线性解码变分自动编码器 (LDVAE),将非线性编码器与线性解码器相结合,以平衡表达能力和可解释性,但也不包含空间信息。SpaGCN、BASS、BayesSpace 和 GraphST 等空间转录组学特定方法相反地采用复杂的模型,例如图神经网络或 Potts 模型来合并空间信息。这些方法提供了灵活性,可以联合分析组学数据和空间信息;然而,这些方法返回的嵌入通常通过聚类和差异表达分析进行事后解释。当前将空间信息纳入降维的可解释方法采用高斯过程或隐马尔可夫随机场,例如 MEFISTO、非负空间分解混合 (NSFH) 和使用矩阵分解 (SpiceMix) 的细胞空间识别等方法。它们提供了空间感知的可解释的降维,但计算成本很高。
在这里,作者提出了用于可解释的空间感知降维的 STAMP。STAMP 基于深度生成模型 prodLDA 构建,该模型通过自动编码和黑盒变分推理确保可扩展性。STAMP 使用简化的图卷积网络作为推理网络,允许在计算成本略有增加的情况下合并空间信息。STAMP 输出一个潜在表示,由空间组织的主题和相关的基因模块组成,其中包含按对主题的贡献排名的基因。这明确地将每个基因的重要性与一个主题联系起来,有助于提高可解释性。如果需要,进一步的分析,例如基因集富集或通路分析,可以为每个主题分配进一步的生物学意义。STAMP 计算每个细胞和每个主题的主题比例分数,每个细胞内的比例总和为 1。对于具有主导主题的细胞,还可以分配与该主导主题相关的解释。作者还在学习的基因模块上引入了结构化的稀疏性先验,使得这些模块在主题之间和主题内都是稀疏的。作者在多个数据集上测试了 STAMP,并表明所学习的主题与组织中已知的解剖结构相对应。这消除了额外的聚类步骤的需要。此外,相关标记基因在随附的基因模块中排名靠前,突出了 STAMP 的可解释性,并提供了差异表达分析的替代方法。
作者将 STAMP 与其他输出非负潜在嵌入及其相应基因模块的降维方法进行了基准测试,并表明 STAMP 在识别生物学相关域及其基因模块方面表现良好。此外,STAMP 在可扩展性方面也优于其他方法。最后,STAMP 是第一个可以跨时间序列空间转录组数据捕获共享主题的主题建模方法。通过小鼠胚胎发育数据集,STAMP 揭示了时空相关的主题及其相关的基因模块。这些主题与已知的生物结构相匹配,相关的基因模块跟踪了贡献基因随时间的变化。
研究结果
1. STAMP 工作流程
STAMP 将主题建模与深度生成模型相结合,从而继承了可解释性和可扩展性的优点。它接收基因表达值及其空间信息,并输出具有相关基因模块的可解释的潜在主题(Fig. 1a,b )。每个相关基因都有一个基因模块分数,表示其对主题的贡献。在处理单个样本的最基本形式中,STAMP 将细胞 中观察到的基因 的表达 建模为从伽玛泊松分布中抽取的样本,该分布由潜在主题 (主题 在细胞 中的比例)、基因模块 (基因 对主题 的贡献)、背景残差 和离散度 的组合确定。为了促进基因模块 中的结构化稀疏性,作者利用结构化正则化马蹄先验。该先验确保每个基因仅涉及主题的子集,并且每个主题仅涉及相关基因模块中有限数量的基因,这有利于模块的生物学解释并为这些模块提供鲁棒性(先验的详细描述在 Methods 部分给出)。为了处理多个样本,作者添加了批次校正项 ,以捕获数据中存在的批次相关变化。最后,作者扩展 STAMP 来处理时间序列数据,通过在 Matern 核之前施加高斯过程,允许基因模块在不同时间点变化。为了将空间信息纳入模型中,作者使用简化图卷积网络(SGCN)作为推理网络,该网络接收基因表达和从空间位置构建的邻接矩阵(Fig. 1a)。通过最大化证据下限 (ELBO),使用黑盒变分推理对整个模型进行端到端训练。模型的详细描述可以在 Methods 和 Supplementary Note 1 中找到。
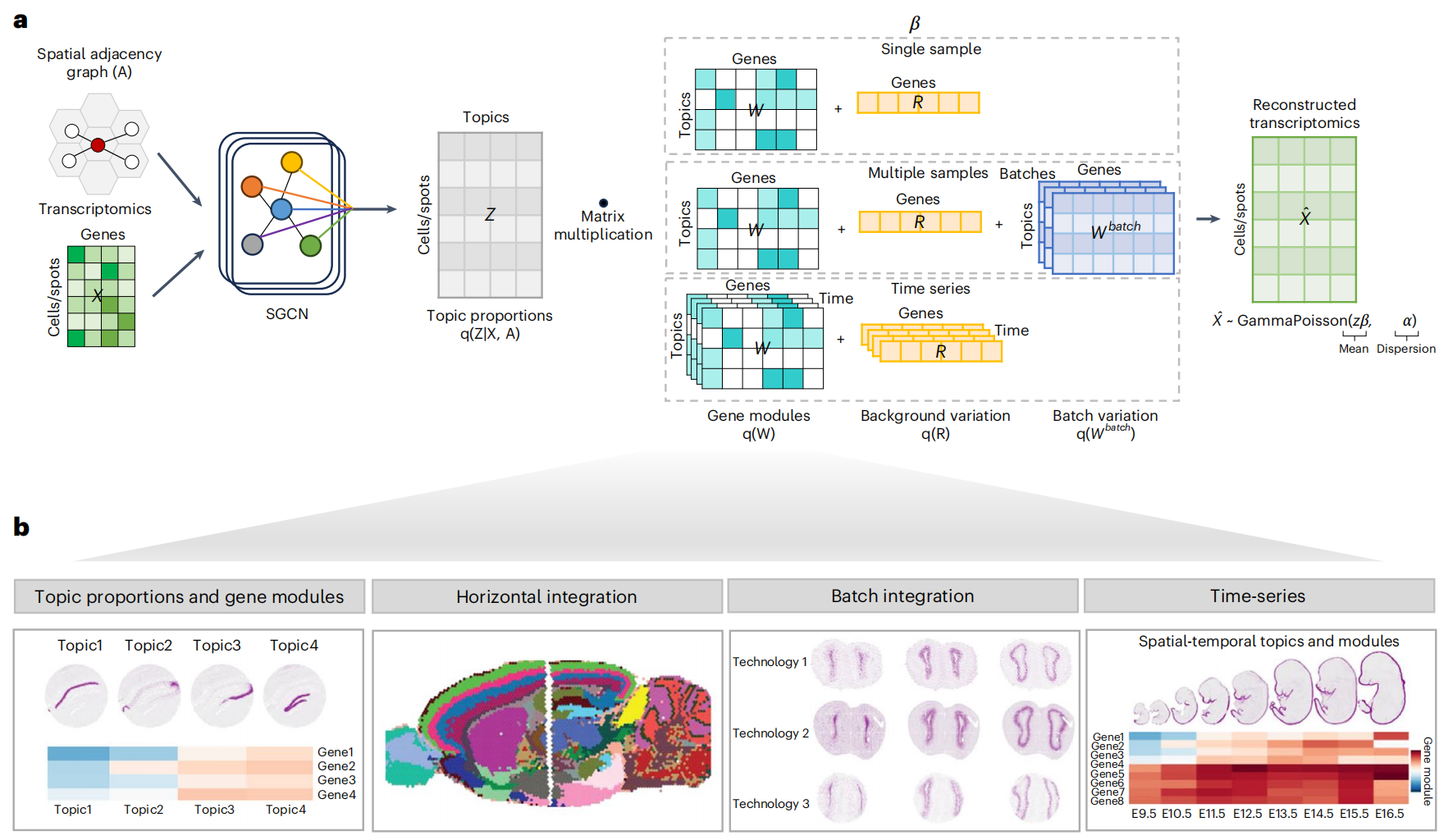 Fig.1 用于空间转录组学分析的可解释的深度生成模型
Fig.1 用于空间转录组学分析的可解释的深度生成模型
a. STAMP 工作流程。STAMP 接收基因表达值和根据点的位置构建的邻域图。STAMP 涵盖三种不同的分析场景,单样本、多样本和时间序列模块。STAMP 使用黑盒变分推理和简化图卷积网络(SGCN)来查找变分参数。
b. STAMP 的功能。基因模块得分 和主题比例 是 STAMP 的输出。这些输出可用于下游分析,例如水平、垂直整合和时间序列分析。
作者首先验证了 STAMP 使用模拟数据集恢复不同层模式的能力。按照 Townes 等人的方法,作者生成了具有五个重叠模式的数据(Extended Data Fig. 1a),其中每个点可以与多个模式相关联。STAMP 能够干净地恢复五种模式(Extended Data Fig. 1a,b)。作者还评估了其他方法,包括 Leiden 聚类、使用 STAGATE、GraphST 和 SpatialPCA 进行空间感知降维,然后是 k-means 聚类。这些基于聚类的分析都无法正确恢复所有模式(Extended Data Fig. 1c)。这证明了 STAMP 主题建模方法在空间重叠模式去卷积方面相对于聚类算法的优势。与将每个点分配给单个 cluster 的传统聚类不同,主题建模为每个点提供概率主题组合,从而允许表示多个身份(主题)。此外,作者还测试了无聚类方法,即 LDA、LDVAE、NMF、NSFH 和 SpiceMix。虽然这些方法的性能优于聚类方法,但它们的效果不如 STAMP(Supplementary Note 2)。
2. STAMP 揭示小鼠海马体的空间域
接下来,作者展示了 STAMP 识别已知组织结构的生物学相关主题和基因模块的能力。作者将 STAMP 应用于使用 Slide-seq V2 生成的包含 39,220 个 spots 的小鼠海马数据集。使用 Allen Brain Atlas 作为解剖区域的 ground truth(Fig. 2a),作者比较了 STAMP 和其他五种方法,NMF、LDA、LDVAE、以及最近为空间转录组学开发的两种方法 NSFH 和 SpiceMix 的性能。所有这些方法都输出非负潜在嵌入及其相应的基因模块。
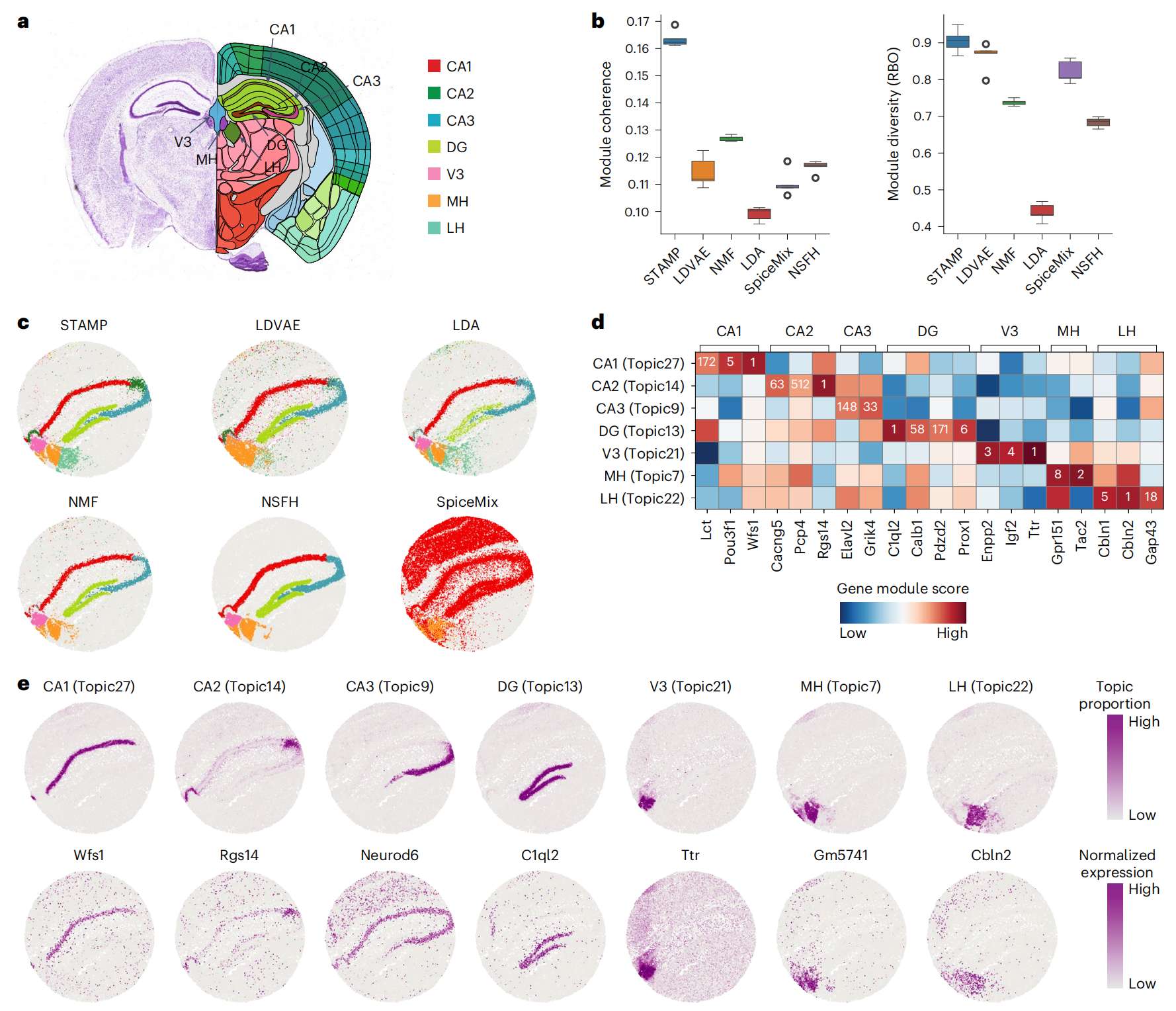 Fig.2 STAMP 准确识别 Slide-seq V2 小鼠海马体中的生物学相关主题
Fig.2 STAMP 准确识别 Slide-seq V2 小鼠海马体中的生物学相关主题
a. Allen 小鼠大脑图谱参考,带注释的海马体区域。
b. STAMP 和 5 种竞争方法在使用不同 seeds 的五次不同运行中获得的模块一致性和模块多样性得分的箱线图。在箱线图中,中心线表示中位数,箱线表示上四分位数和下四分位数,须线表示 1.5 × 四分位数范围。
c. 通过这些方法捕获的与海马体相关的选定空间主题。为了便于可视化,主题被二值化。
d. 不同海马区域已知基因标记的基因模块排名(数量)和分数(颜色)。
e. 通过 STAMP 识别的空间主题以及每个基因模块中相应最高排名基因的表达水平的可视化。
作者首先根据主题相关基因模块量化了 STAMP 和竞争方法的性能(Fig. 2b)。作者在主题建模文献中采用了两个常用的指标:模块一致性和模块多样性。模块一致性衡量模块内排名靠前的基因之间的共表达程度,而模块多样性衡量基因模块的独特性。STAMP 在这两个指标中得分最高,模块一致性中值为 0.162,模块多样性为 0.9。所有其他方法在相干性度量方面的得分都要差得多,其中 NMF 排名第二(0.127)。在多样性方面,LDVAE 得分第二(0.87),而在模块一致性方面则较差(0.11)。LDA 表现最弱,在模块一致性 (0.1) 和多样性 (0.44) 方面得分垫底。
STAMP 输出的视觉检查显示了明确的空间主题,对应于海马体的七个关键解剖区域,即 Cornu Ammonis 1 (CA1)、CA2、CA3、齿状回 (DG)、第三脑室 (V3)、内侧缰核 (MH)和外侧缰核(LH)(Fig. 2c and Extended Data Fig. 2)。相比之下,没有其他方法能够分离 CA2 和 CA3 区域,也无法正确捕获 LH 区域。LDA、NMF 和 NSFH 正确捕获 CA1、DG、V3 和 MH,但未能捕获 LH 并分离 CA2 和 CA3。LDVAE 捕获了 CA1 和 DG,但没有捕获 LH,并将 CA2 与 CA3 合并,以及 V3 与 MH 合并。SpiceMix 的表现最差,未能捕获任何解剖区域(Extended Data Fig. 2 and Supplementary Figs. 1--6)。作者通过检查已发表的解剖区域基因标记的排名进一步研究了各自的基因模块(Fig. 2d )。对于除 CA3 之外的所有区域,在前 10 名中至少发现一个标记(CA1:Wfs1、Pou3f1;CA2:Rgs14;DG:C1ql2、Prox1;V3:Enpp2、Igf2、Ttr;MH:Tac2、Gpr151;LH:Cbln1、Cbln2)。这些各自解剖区域的成熟标记基因提供了证据,表明 STAMP 可以生成具有生物学意义的主题和基因模块。然后,作者可视化了 STAMP 主题比例和各个模块中顶级基因表达的空间分布,确认主题比例和基因表达水平是一致的(Fig. 2e)。
3. STAMP 揭示了癌症相关成纤维细胞的主题
接下来,作者使用 CosMx SMI 获取的人类非小细胞肺癌 (NSCLC) 数据集中的一个样本评估了 STAMP 揭示细胞状态和相关基因模块的能力。选定的肺 #5-3 样本由 93,206 个 spots 组成,捕获了 960 个基因(Fig. 3a )。这里作者设置 STAMP 返回 15 个主题及其基因模块。使用 DISCO 的 scEnrichment 工具对基因模块进行分析,以注释当前的主题(Supplementary Table 1)。作者发现主题注释与原始研究中手动注释的细胞类型非常吻合,证实了主题及其基因模块在捕获细胞状态方面的准确性(Extended Data Fig. 3a,b)。接下来,作者绘制了已知细胞类型标记的基因模块排名,以验证基因模块的生物学相关性(Fig. 3b)。大多数细胞类型标记物在各自的基因模块中排名靠前,至少有一个标记物位于前 10 名(肿瘤边缘除外),大多数位于前 20 名,证实了基因模块的生物学相关性。作者还可视化了每个主题的 top 基因的空间表达以及前 20 个基因的聚合表达。这些基因表现出高空间共表达模式,并与各自的主题准确相关,突出了基因模块与其主题之间的一致性(Supplementary Fig. 7)。
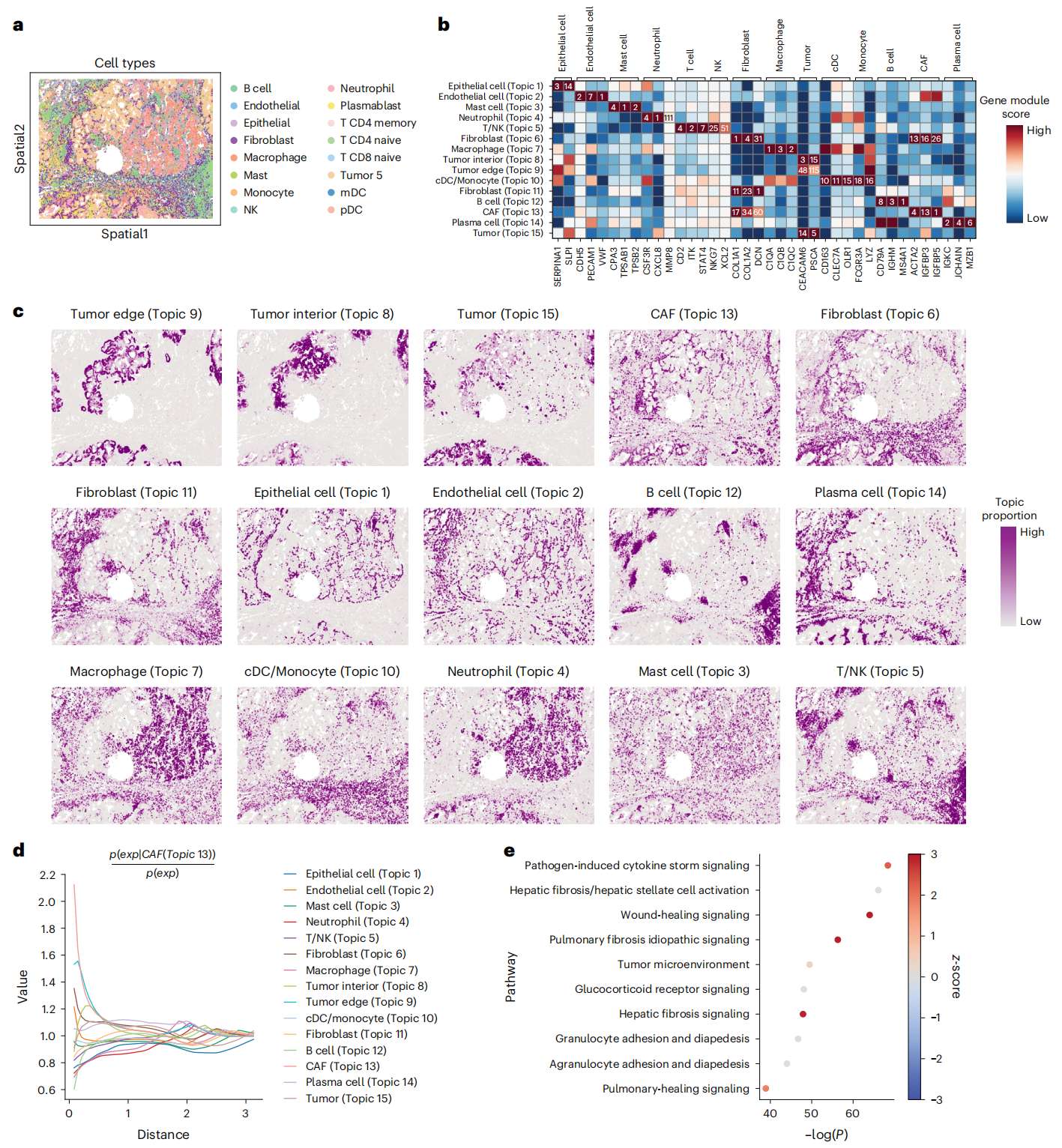 Fig.3 STAMP 对 SMI NSCLC 数据中的常规成纤维细胞中的 CAFs 进行反卷积
Fig.3 STAMP 对 SMI NSCLC 数据中的常规成纤维细胞中的 CAFs 进行反卷积
a. 使用 CosMx 空间分子成像仪 (SMI) 获取带有原始细胞类型注释的 NSCLC 样本数据。
b. 每个主题的标记基因的基因模块排名(数量)和分数(颜色)。
c. 使用其基因模块注释的主题的空间图。
d. 使用 Squidpy 计算的不同细胞类型相对于 CAFs 的空间共现(Topic 13)。CAFs 最靠近肿瘤边缘(Topic 9)。
e. 从 CAFs 针对其他成纤维细胞群的独创性途径分析 (IPA) 中获得的规范途径富集分析。点的颜色反映了 CAFs 与成纤维细胞的 IPA z-score 富集,红色表示预测的通路激活,蓝色表示通路抑制。x 轴通过 -log10(adj. P) 显示显着性水平。Adj. P values 通过 Fisher 精确检验(right tailed)计算,然后进行 Benjamini-Hochberg 调整。NK, natural killer; cDC, conventional dendritic cell; mDC, myeloid dendritic cell; pDC, plasmacytoid dendritic cell。
STAMP 的主题还以比原始研究更高的分辨率实现了细胞类型注释(STAMP in Fig. 3c , original in Extended Data Fig. 3a, comparison in Extended Data Fig. 3b)。例如,STAMP 将肿瘤分为三个区域:肿瘤(Topic 15)、肿瘤内部(Topic 8)和肿瘤边缘(Topic 9),并得到标记基因的支持。成纤维细胞还以更高分辨率分解为两个成纤维细胞主题和一个癌症相关成纤维细胞(CAFs)。两个成纤维细胞主题具有不同的特征,其中 Topic 6 成纤维细胞中的胶原基因(例如 COL3A1 和 COL1A1)和 Topic 11 成纤维细胞中的蛋白多糖基因(例如 DCN、MMP2 和 LUM)表达较高,代表不同的成纤维细胞亚群(Extended Data Fig. 3c)。胶原蛋白通过形成构成主要组织结构的纤维束,构成结缔组织细胞外基质 (ECM) 的大部分,而蛋白聚糖是可溶性糖蛋白,与胶原纤维形成大型复合物。作者还根据报告的 CAF 特征基因(包括 PDGFRB、ACTA2、MYH11、IGFBP5 和 IGFBP3)注释了 CAFs(Topic 13)(Supplementary Table 1 and Fig. 3b)。作者发现 CAF topic 集中在肿瘤边缘的外部,这与 CAFs 形成将肿瘤组织与其他基质区域分开的屏障的报道相匹配。因此,作者通过使用 Squidpy 计算主题的共现概率来量化主题之间的空间关系。计算出的关于 CAF topic 的共现概率表明它与肿瘤边缘具有最高的共现概率,从而进一步加强了作者分配的注释(Fig. 3d)。
由于 CAFs 对肿瘤微环境产生重要影响,从而促进炎症和肿瘤进展,因此作者对其中的分子途径很感兴趣。因此,作者通过计算其相对于其他成纤维细胞的差异表达基因(Topics 6 and 11)来表征 CAF topic,然后进行通路分析(Fig. 3e)。分析显示细胞因子信号传导上调,这可归因于肿瘤组织内分泌的炎症分子。纤维化途径也上调,表明 CAFs 与肺纤维化途径的发展之间的分子机制可能存在重叠。最后,伤口愈合途径也被上调,据报道,这有助于上皮肿瘤发育过程中基质组织的形成,并参与促肿瘤细胞与肿瘤细胞的串扰。
4. STAMP 缝合小鼠前脑和后脑切片
由于采集技术的限制,需要单独的实验来从同一切片中捕获更大区域的组织。为了分析此类多样本数据,作者在模型中加入了一个额外的潜在变量,以创建具有批次校正功能的 STAMP(Fig. 1a )。作者首先使用小鼠大脑矢状切片的两个 10x Genomics Visium 数据集(分为后部和前部)证明了 STAMP 发现不同样本中共同主题的能力(Fig. 4a )。在这里,作者将 STAMP 与 NMF、LDA、LDVAE、NSFH 和 SpiceMix 进行基准测试,通过指标和目视检查对其进行评估。作者首先量化了它们在基因模块一致性和多样性方面的表现(Fig. 4b )。STAMP 在模块多样性和模块一致性方面都表现出了顶尖的性能。LDVAE 在这两个指标中均排名第二。与小鼠海马体的例子一样,LDA 在这两个指标中都取得了最差的表现。使用 Allen Brain Atlas 作为参考(Fig. 4c ),作者直观地比较了 STAMP 和竞争方法的输出(Fig. 4d and Supplementary Figs. 8--13)。总体而言,所有方法都能够发现两个部分之间一致的主题,但并非所有方法都与参考文献相匹配。STAMP 能够清晰地捕获大脑皮层、海马体(CA 和 DG)和小脑的内部结构,并分离尾状核(CP)和伏隔核(ACB)。在竞争方法中,只有 LDA 和 NMF 能够分离一些上皮质层,而只有 NMF 和 NSFH 捕获海马中的 CA 和 DG 并分离 CP 和 ACB。相反,除 LDA 之外的所有方法都捕获了小脑的结构。
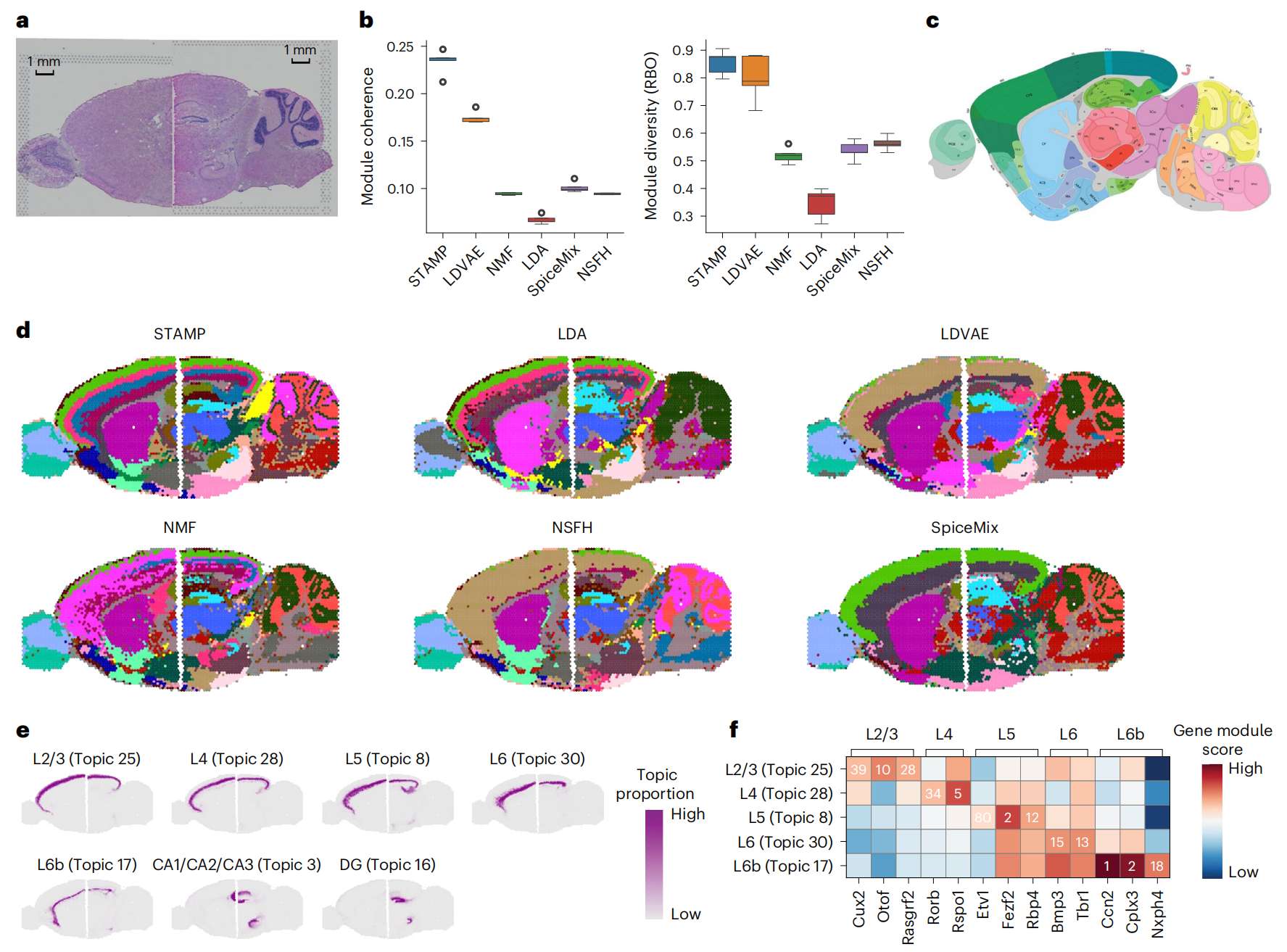 Fig.4 STAMP 描绘了多个 Visium 小鼠脑组织切片的不同皮质层
Fig.4 STAMP 描绘了多个 Visium 小鼠脑组织切片的不同皮质层
a. 小鼠大脑前部和后部切片的苏木精和伊红 (H&E) 图像。
b. STAMP 和 5 种竞争方法在使用不同 seeds 的五次不同运行中获得的模块一致性和模块多样性得分的箱线图。在箱线图中,中心线表示中位数,箱线表示上四分位数和下四分位数,须线表示 1.5 × 四分位数范围。
c. 来自 Allen 小鼠大脑图谱的带注释的小鼠大脑矢状切片图像。
d. STAMP 和 5 种竞争方法返回的空间主题。每个主题都被二值化,以便于在单个图中可视化。
e. STAMP 专门识别的大脑皮层层。
f. 大脑不同层中已知基因标记的基因模块排名(数量)和分数(颜色)。
作者进一步研究了 STAMP 的主题及其相关基因模块,重点关注皮质层和海马体(Fig. 4e )。STAMP 能够识别两个大脑部分共享的主题并将它们很好地对齐。为了验证各层是否被正确捕获,作者检查了报告的标记在各自基因模块中的排名(Fig. 4f)。作者发现大多数标记都排名前 20 名,并且三层中有一个标记排名前 5 名。这肯定了 STAMP 模型捕获具有生物学意义的主题和基因模块的能力。
5. STAMP 识别不同技术之间的共享主题
在这里,作者展示了 STAMP 从使用不同技术获取的多个数据集中恢复主题和基因模块的能力。在这里,作者使用了三个小鼠嗅球(Fig. 5a )数据集,每个数据集均通过不同的技术,Slide-seq V2、Stereo-seq 和 10x Genomics Visium 获得,总共 148,087 个细胞。从使用 PCA 生成的 UMAP 可视化结果来看,原始数据中存在显着的批量效应(Fig. 5b)。作者对 STAMP 以及 LDA、LDVAE、NMF、NSFH 和 SpiceMix 进行了基准测试,以评估数据集成性能。作者添加了另外三种算法:DeepST、PRECAST 和 STAligner,它们提供了无法解释的嵌入,但能够纠正空间数据中的批次效应。由于内存限制,SpiceMix、DeepST 和 STAligner 方法无法在这些数据集上生成结果。
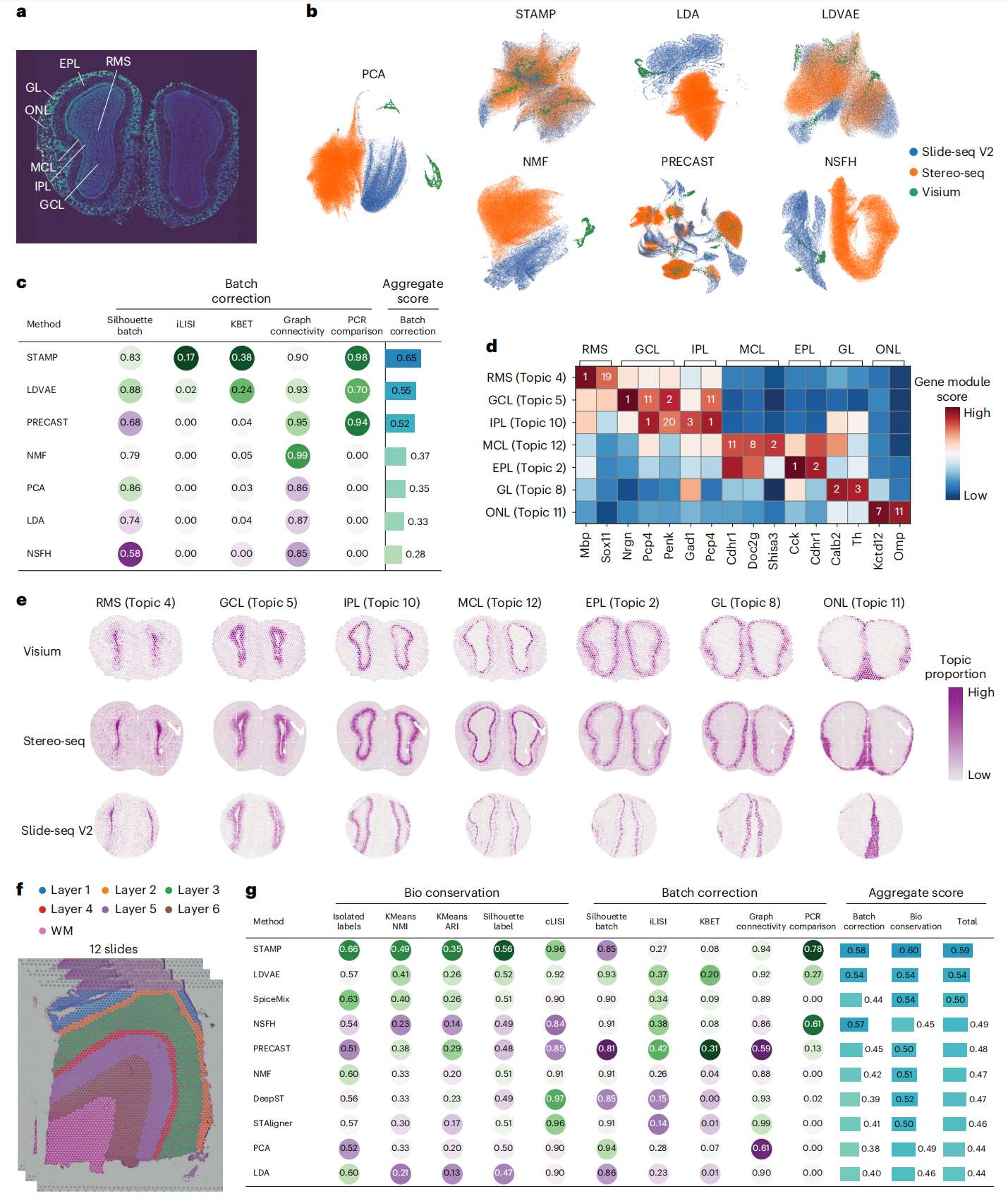 Fig.5 STAMP 消除批次效应
Fig.5 STAMP 消除批次效应
a. 带注释的 DAPI 染色图像显示了小鼠嗅球的层状组织。
b. 使用每个方法返回的潜在空间/主题计算 UMAP。UMAP 根据各自的技术进行着色。
c. 表格显示了为每种方法计算的 scIB 指标。得分为 1 表示最佳性能。
d. STAMP 针对每个主题返回的标记基因的基因模块排名(数量)和分数(颜色)。
e. 在 10x Genomics Visium、Stereo-seq 和 Slide-seq V2 数据中识别出不同嗅觉层的主题。
f. 带有原始注释的 12 个 DLPFC 切片的数据集。
g. 表格显示了为每种方法计算的 scIB 指标和聚合分数。得分为 1 表示最佳性能。
作者使用所有方法将 12 个主题拟合到数据中,并使用 UMAP 可视化它们的输出。在 STAMP 的输出中,三个数据集高度混合(Fig. 5b )。对于 LDVAE 和 PRECAST 可以观察到一些混合,但仍然可见大量特定于批次的 clusters。对于 LDA、NMF 和 NSFH,数据集以特定于批次的方式保持分离,因为它们没有明确设计用于处理批次效应。作者进一步量化了与 scIB 指标(silhouette score, iLISI, kBET, graph connectivity and PCR comparison)的数据集成,并且汇总结果(Fig. 5c)与 UMAP 图大致匹配。STAMP 获得了最好的总分,LDVAE 排名第二,其次是 PRECAST、NMF、LDA 和 NSFH,其总分与 PCA 输出相当,这意味着数据集成较差。
通过检查 STAMP 主题的相关基因模块,作者发现了与小鼠嗅球已知层相关的基因,从而可以进行手动注释(Fig. 5d and Supplementary Fig. 14)。作者能够识别头端迁移渠道(RMS)、颗粒细胞层(GCL)、内网状层(IPL)、僧帽细胞层(MCL)、外网状层(EPL)、嗅小球层(GL)和嗅神经层(ONL)。许多报告的标记在其模块中排名靠前,即 Mbp(在 RMS 中排名第一)、Nrgn 和 Penk(在 GCL 中分别排名第一和第二)、Pcp4 和 Gad1(在 IPL 中分别排名第一和第三)、Shisa3 和Doc2g(在 MCL 中分别排名第二和第八)、Calb2(在 GL 中排名第二)和 Kctd12(在 ONL 中排名第七)。值得注意的是,GCL 和 IPL 共享许多上调基因,例如 Pcp4 和 Penk。作者还可视化了三个数据集中主题的空间分布(Fig. 5e ),并发现它们根据嗅球的结构正确组织(Fig. 5a)。这清楚地证明了 STAMP 能够识别通过不同技术获取的数据中的共享主题,并纠正大量批次效应。
接下来,作者对使用 10x Genomics Visium 采集的 12 个背外侧前额叶皮层 (DLPFC) 切片的集合运行 STAMP(Fig. 5f )。原始数据被注释以指示皮质层和白质。这种 ground truth 注释的可用性使作者能够测试生物特征的保守性(生物保守性),同时通过相对大量的批次最小化批次效应(批次校正)。在这里,作者用 STAMP 和竞争方法拟合了 10 个主题。使用 UMAP 可视化集成输出,除 PRECAST 之外的所有方法都能够保持皮层层和白质之间的分离(Extended Data Fig. 4a)。所有方法都不同程度地混合批次,但批次特定区域在 DeepST 和 PRECAST 输出中高度可见(Extended Data Fig. 4b)。对于定量评估,作者采用了五个用于生物保护的指标(isolated labels, k-means-based NMI, k-means-based ARI, silhouette score for annotation labels and cLISI)和五个用于批次校正的指标(silhouette score for batch, iLISI, kBET, graph connectivity and PCR comparison)来自 scIB 指标。在总评分中,STAMP 在生物保守性和批次校正方面均名列前茅(Fig. 5g)。对于生物保守性,STAMP 在所有指标中均名列前茅,但 cLISI 除外,它仅落后于 DeepST,仅略有差距(0.96 比 0.97)。STAMP 还能够捕捉到其主题的清晰分层模式(Supplementary Fig. 19)。
6. STAMP 揭示胚胎发育的时空主题
最后,作者证明了 STAMP 从时间序列空间转录组数据中发现时空相关主题和相关基因模块的能力。作者分析了从 E9.5 到 E16.5 八个时间点的小鼠胚胎样本的 Stereo-seq 数据,总数据集大小超过 540,000 个细胞(Fig. 6a )。为了解释基因表达的动态变化,允许基因模块通过高斯过程随时间点变化。这种动态表述使 STAMP 能够捕捉跨时间的相关主题。作者拟合了 40 个主题,捕捉不同时间点的匹配组织,说明它们的发育轨迹(Extended Data Fig. 5a)。二值化主题(Extended Data Fig. 5b)解析了与数据集的原始聚类和注释相匹配或超出的生物学细节(Fig. 6a)。值得注意的是,STAMP 主题的空间平滑度可以通过调整 SGCN 的层数来控制,层数越高,主题越平滑。作者用不同数量 SGCN 层的 E11.5 胚胎切片获得的结果证明了这一点,并使用 Moran's I 指数定量地证明了主题空间平滑度的变化(Extended Data Fig. 6a,b)。
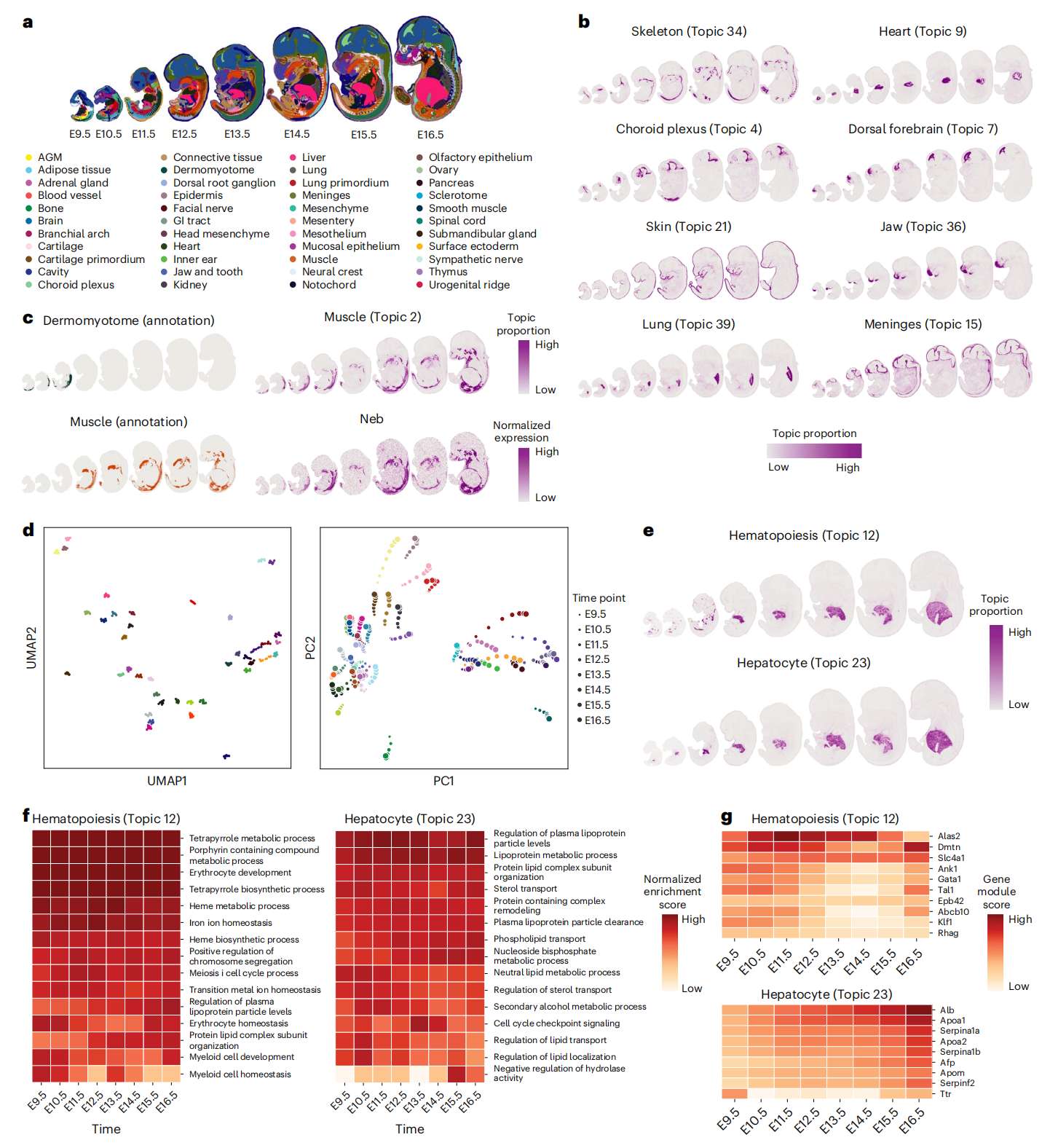 Fig.6 Stereo-seq 胚胎数据的时间序列分析
Fig.6 Stereo-seq 胚胎数据的时间序列分析
a. 小鼠胚胎发育从 E9.5 到 E16.5 的 Stereo-seq 数据系列,带有原始注释。
b. STAMP 识别了骨骼、心脏、皮肤、下颌、脉络丛、背侧前脑、肺和脑膜等时空模式。所有拟合的主题都可以在 Extended Data Fig. 5 中找到。
c. 原始注释(左)和 STAMP 输出(Topic 2)的比较。STAMP 将生皮肌节和肌肉区域合并为一个主题。NEB 的空间基因表达(从相关基因模块中识别)支持合并。
d. STAMP 输出的 UMAP 和 PCA 图揭示了空间连接的基因模块的连续轨迹。
e. STAMP 确定的肝细胞和造血主题。
f. 使用肝细胞和造血主题的基因模块计算的 GSEA 标准化富集分数。选择并显示按标准化富集分数排序的前 15 个通路。
g. 肝细胞和造血主题中选定标记的基因模块分数。AGM, aorta-gonad-mesonephros; GI, gastrointestinal。
与 STAMP 主题相关的组织包括主要器官,如骨骼、肝脏、心脏、皮肤、下颌和肺。在颅骨区域内,STAMP 分解了脉络丛和前脑等组织(Fig. 6b )。通过检查各个主题,还可以观察主题随时间的对应关系,跟踪组织发育轨迹(Fig. 6b )。一个例子是心脏(Topic 9)变大,作者对 E11.5 的结果表明心室的形成。另一个例子是在 E9.5 开始发育的皮肤(Topic 21),到 E13.5 我们可以看到包裹胚胎的可辨别结构。对于脑膜(Topic 15),它在整个发育过程中保持一致的结构形式。此外,STAMP 可以绘制发育连续体图,将不同的早期和晚期形式联系起来,这些形式通常被传统的聚类分析识别为不相关的聚类。例如,STAMP 在单个主题(Topic 2)中捕获了从生皮肌节到完全生长肌肉的连续肌肉发育,而原始注释将其显示为两个不同的 clusters(Fig. 6c)。值得注意的是,相关基因模块包含 Neb,这是一种重要的骨骼肌发育标志物,其表达与主题具有相同的空间分布,从而支持合并。
相关的基因模块还反映了每个主题在时间点之间的相似性以及随时间的逐渐变化。在 UMAP 可视化中,同一主题的基因模块紧密聚集在一起(Fig. 6d , left)。通过 PCA,同一主题的基因模块也聚集在一起。值得注意的是,主要成分还捕捉了许多主题随时间的渐进变化(Fig. 6d, right)。这些有序且渐进的变化凸显了 STAMP 使用基因模块对其进行建模的能力。他们还说明了组织之间共享的开发程序,并且可以进一步利用基因模块来获得进一步的生物学见解。
为了显示连接的基因模块捕获的生物信息,作者重点关注两个重要主题:造血(Topic 12)和肝细胞(Topic 23),它们共定位于发育中的肝脏(Fig. 6e )。在胚胎发育过程中,早期发育过程中,造血发生在卵黄囊,然后从接种的造血干细胞转移到肝脏。到 E12.5,肝脏是造血的主要场所,然后到 E15.5 逐渐转移到附近的脾脏。E16.5 之后开始进一步迁移到骨髓。因此,造血和肝细胞主题在空间上与 E12.5 几乎完全重叠;然而,它们的基因模块清楚地将它们分开。使用基因集富集分析(GSEA),肝细胞主题的基因模块显示出脂质加工和其他代谢过程的强烈富集,而与造血过程相关的基因集,特别是红细胞和血红蛋白相关的代谢过程,在造血主题中具有高度代表性(Fig. 6f )。作者进一步证实,红细胞和肝细胞的标记物在造血和肝细胞主题中分别具有较高的基因模块得分(排名在前 100 名内)(Fig. 6g)。
讨论
在这项工作中,作者开发了 STAMP,一种用于识别空间转录组数据中的主题和相关基因模块的深度概率方法。为了合并空间信息,作者将根据空间位置构建的邻接矩阵提供给 STAMP,后者使用 SGCN 对空间邻接进行编码。STAMP 还在基因模块上采用了正则化的马蹄形先验,以鼓励结构化稀疏性,从而产生更稳健和可解释的估计。作者在多个数据集上测试了 STAMP,与 NMF、LDA、LDVAE、NSFH 和 SpiceMix 等其他竞争方法相比,在模块一致性和多样性方面取得了良好的性能。在小鼠海马数据集中,STAMP 识别了与海马内不同解剖区域相关的不同基因模块,捕获了该大脑区域功能多样性背后的分子异质性。通过人类肺癌数据集,STAMP 捕获了与特定肿瘤区域和细胞类型相对应的空间组织基因表达模式。这使得能够以比原始研究更高的分辨率进行注释,将肿瘤分为不同的区域,例如肿瘤边缘和肿瘤内部,并解析其他成纤维细胞子集,例如 CAFs。
作者还扩展了 STAMP 以集成多个数据集甚至时间序列数据。作者对两个小鼠大脑矢状切片(前部和后部)进行了整合分析,捕获了生物学上准确的主题,这些主题也沿着两个切片之间的共享边缘对齐。STAMP 还展示了将从不同技术和批次获取的数据与小鼠嗅球切片以及人类 DLPFC 切片的数据整合的能力。
最值得注意的是,作者使用 STAMP 在八个时间点的一系列发育中的小鼠胚胎切片中对时空相关主题进行建模。与原始研究的注释相比,STAMP 以更高的分辨率揭示了复杂的解剖结构。通过根据高排名基因和富集途径分析相关基因模块,作者注释了更高分辨率的主题并揭示了它们的生物学意义。例如,STAMP 准确捕获了肝脏和造血主题,相关基因在其相关基因模块中排名靠前。值得注意的是,在胚胎肝脏是造血主要场所的时期,造血和肝脏主题在空间上是一致的。这些结果凸显了空间转录组学的力量以及 STAMP 在破译复杂生物系统中的效用。
作者还注意到 STAMP 的一些局限性和未来发展的领域。在目前的形式下,STAMP 无法促进不同条件之间的比较分析,例如正常与癌症或突变与野生型。此类分析对于健康和疾病相关的研究非常重要,可以捕捉和剖析详细的表型差异,并可能导致机制解释。因此,未来的发展是修改 STAMP 以恢复不同条件下不同的主题。此外,进一步的发展是可选择包含先前的知识作为输入,例如基因集或途径、细胞类型或生态位信息,这些信息可能部分可用于感兴趣的组织。这可以帮助指导利用先前的生物学知识构建基因模块,并有可能实现更好的准确性和生物学相关性。最后,进一步发展的另一个途径是扩展 STAMP 以处理空间多组学数据或图像数据,包括镶嵌数据场景,其中并非所有数据集都具有相同的可用数据模式。包含额外的数据模式增加了信息内容捕获,包括正交信息,从而提高了整体准确性。
作者设计的 STAMP 是用户友好的,并且能够处理来自不同实验平台的数据。STAMP 可以扩展到包含数十万个细胞的非常大的数据集,其中测试的最大数据集包含超过 500,000 个细胞,确保其随着数据集大小的增长而相关(Extended Data Fig. 7)。所有分析均在配备 Intel Core i7-8665U CPU 和 12GB 内存的 NVIDIA Titan V GPU 的服务器上进行。
--------------- 结束 ---------------
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。

本文由mdnice多平台发布