rm(list = ls())
# 加载必要包
library(data.table)
library(circlize)
library(ComplexHeatmap)
library(rtracklayer)
library(GenomicRanges)
library(BSgenome)
library(GenomicFeatures)
library(dplyr)
### 数据准备阶段 ###
# 1. 读取染色体长度信息
df <- read.table('all_noContig.sizes', col.names = c('chr_id','chr_len'))
# 2. 读取基因组序列
genome <- readDNAStringSet("TG1.LG.fasta")
# 3. 计算GC含量(窗口:10kb,步长5kb)
window_size <- 10000
step <- 5000
gc_content <- lapply(names(genome), function(chr) {
seq <- genome[[chr]]
starts <- seq(1, length(seq) - window_size, by = step)
ends <- starts + window_size
gc <- sapply(1:length(starts), function(i) {
subseq <- subseq(seq, starts[i], ends[i])
sum(alphabetFrequency(subseq)[c("C","G")]) / window_size
})
data.frame(chrom = chr, start = starts, end = ends, gc = gc)
}) %>% bind_rows() %>%
filter(!grepl("Contig", chrom)) %>%
setNames(c('chr_id','bin_start','bin_end','gc'))
# 4. 读取基因密度数据
gc <- read.table('gene_counts.txt',
col.names = c('chr_id','bin_start','bin_end','gene_count')) %>%
filter(!grepl("Contig", chr_id))
# 5. 处理CDS注释数据
txdb <- makeTxDbFromGFF("TG1.gene.new.gff")
cds_ranges <- cds(txdb) %>%
as.data.frame() %>%
dplyr::select(seqnames, start, end) %>%
setNames(c('chr_id','bin_start','bin_end'))
pdf('circle.pdf')
### 绘图参数初始化 ###
circos.clear()
col_text <- 'grey20'
# 关键参数设置:统一轨道高度与边距
circos.par(
gap.degree = 5, # 染色体间空隙
start.degree = 86, # 起始角度
track.height = 0.15, # 统一轨道高度比例
track.margin = c(0.01, 0.01), # 垂直边距压缩
cell.padding = c(0,0,0,0),
clock.wise = TRUE
)
# 初始化染色体布局
circos.initialize(
factors = df$chr_id,
xlim = cbind(rep(0, nrow(df)), df$chr_len)
)
### 绘图轨道绘制 ###
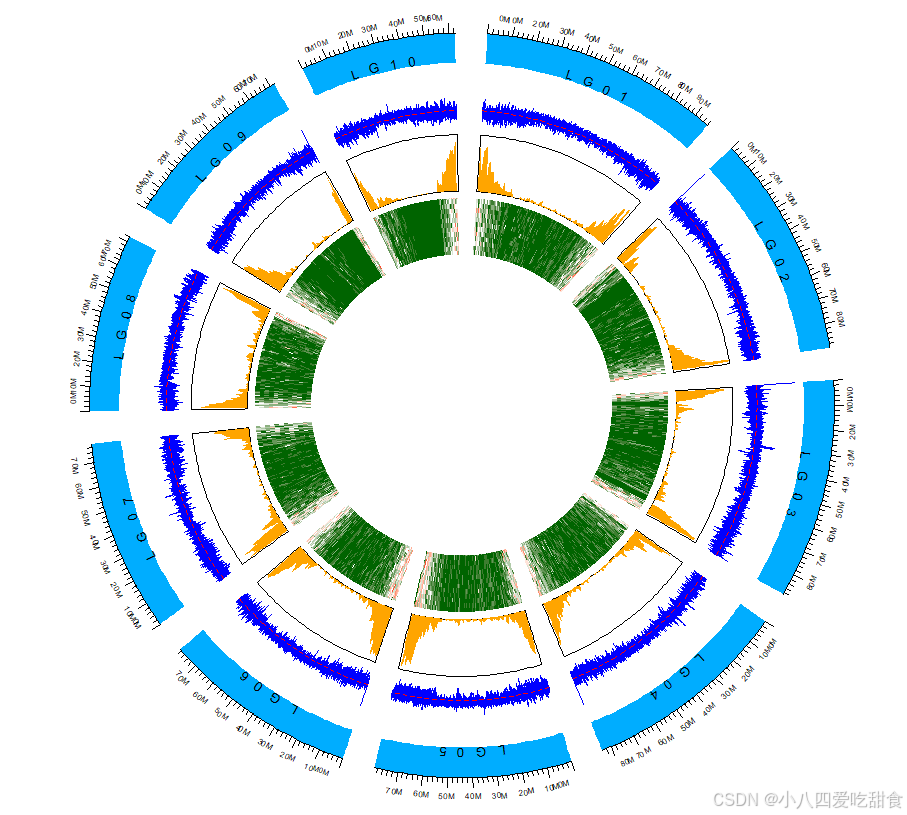
# 轨道1:染色体名称
circos.track(ylim = c(0, 1), panel.fun = function(x, y) {
chr = CELL_META$sector.index
circos.text(CELL_META$xcenter, CELL_META$ycenter, chr,
facing = "bending.inside", cex = 0.8)
}, bg.col = "#00ADFF", track.height = 0.08, bg.border = NA)
# 轨道2:刻度标签
brk <- seq(0, 100e6, by = 10e6)
circos.track(track.index = get.current.track.index(),
panel.fun = function(x, y) {
circos.axis(h = "top", major.at = brk,
labels = paste0(brk/1e6, "M"),
labels.cex = 0.5)
}, bg.border = NA)
# 轨道4:GC含量
circos.genomicTrack(gc_content, panel.fun = function(region, value, ...) {
circos.genomicLines(region, value, col = "blue", lwd = 0.5)
circos.lines(CELL_META$cell.xlim, rep(mean(value[[1]]), 2),
col = "red", lty = 2)
}, track.height = 0.15, bg.border = NA)
# 轨道5:CDS密度
circos.genomicDensity(cds_ranges, col = c("orange"),
bg.border = NA,
track.height = 0.15, window.size = 1e6)
# 轨道3:基因密度
color_genes <- colorRamp2(c(0, 6, 13), c("darkgreen", "white", "red"))
circos.genomicTrack(gc, panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_genes(value[[1]]),
border = NA)
}, track.height = 0.15, bg.border = NA)
dev.off()
#
### 图例添加 ###
lgd_list <- list(
Legend(col_fun = color_genes, title = "Gene Density"),
Legend(labels = c("GC Content", "Genome Average"),
type = "lines",
legend_gp = gpar(col = c("blue", "red"), lty = c(1,2))),
Legend(col = "orange", title = "CDS Density", type = "points")
)
draw(lgd_list, x = unit(0.85, "npc"), just = "left")
### 图像输出 ###
circos.clear()