StructGuy:破解蛋白变异预测的数据泄漏难题
目录
- StructGuy 通过严格避免训练数据泄漏,利用梯度提升树和结构特征,实现了对未见蛋白变异功能效应的精准预测与机制解释。
- 现有细胞基于主体模型(ABM)过于僵化,阻碍复杂生物系统模拟。我们需要转向模块化「积木式」构建,像搭建神经网络般灵活组装模型。
- FoldSAE 利用稀疏自编码器解析 RFdiffusion 的内部机制,实现对蛋白质二级结构的精细调控,将随机生成转变为可控工程。
- 结合机器学习与分子动力学模拟,该研究揭示了锁定 EGFR L858R/T790M 突变体失活态的机制,筛选出 10 个预测效力优于 EAI001 的潜在变构抑制剂。
- AlphaFold3 在 TCR-pMHC 复合物结构预测中确立了领先地位,其 CDR3 区域的置信度分数成为判断结合质量与突变亲和力的核心指标。
1. StructGuy:破解蛋白变异预测的数据泄漏难题
基于 AI 的蛋白质变异效应预测存在一个普遍问题:数据泄漏。许多模型在测试集上表现优异,是因为训练时接触过相似的序列或同源蛋白。一旦面对训练集中未出现过的全新蛋白质,这些模型的表现就会急剧下降,这在创新靶点药物研发中难以接受。
StructGuy 旨在解决这个问题。
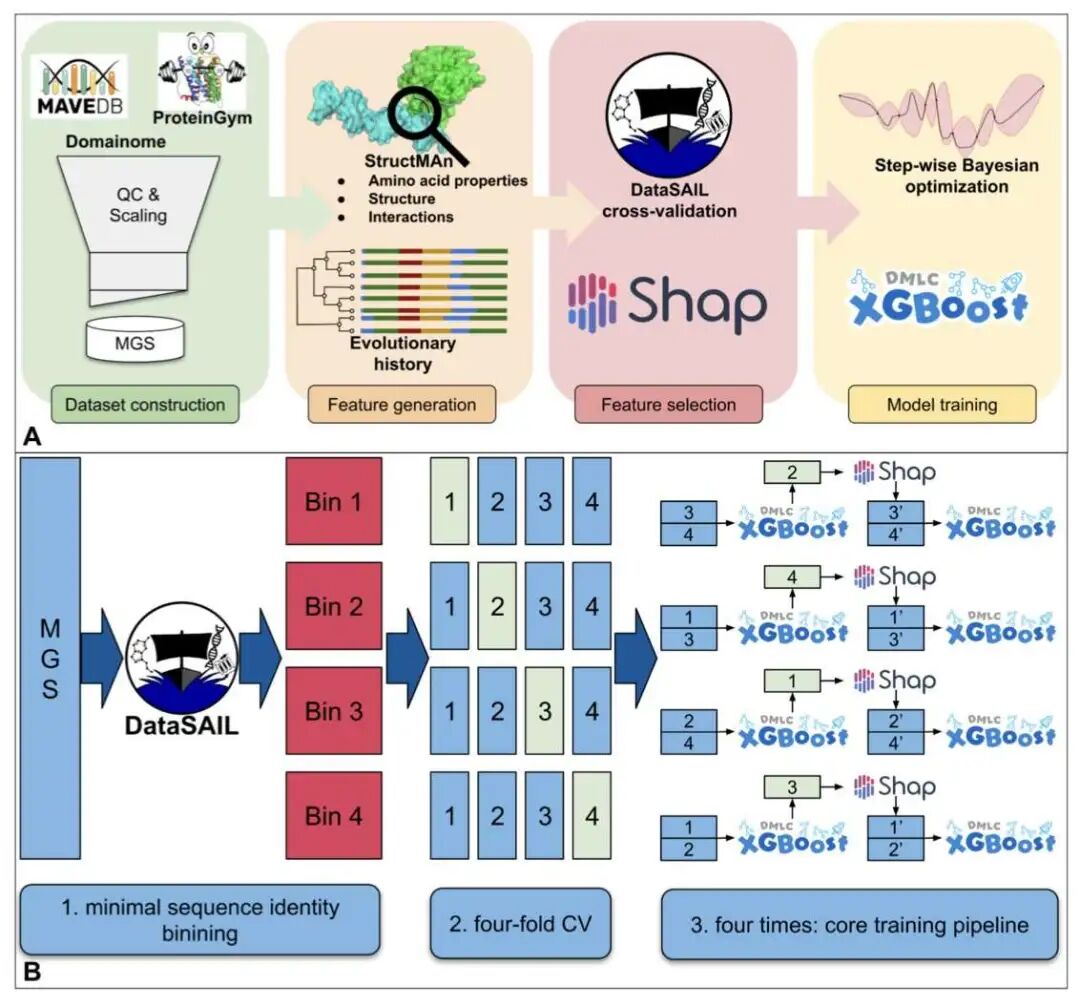
研究者将重点放在了数据的「纯净度」上。他们构建了一个基于 MAVE (多重变异效应分析) 实验的专用数据集,并对其进行严格清洗和分割,确保训练集和测试集之间没有重叠。这样训练出的模型,才真正具备对未知蛋白质的泛化能力。
算法上,StructGuy 选择了梯度提升树 (Gradient Boosting Trees)。它整合了全面的蛋白质结构特征(包括残基相互作用)和进化信息,使预测结果变得完全可解释。用户不仅能得知某个突变有害,还能理解其具体原因,例如破坏了哪个氢键或影响了哪段构象。
研究团队在一个修改版的 ProteinGym 基准测试上验证了该模型。结果显示,StructGuy 的准确度可与目前最先进的零样本(Zero-shot)方法相媲美。
以PPARG (过氧化物酶体增殖物激活受体伽玛) 为例,这是一个重要的代谢类药物研发靶点。StructGuy 在此案例中准确预测了突变的功能影响,并从分子层面提出了机制假设。这种能力对药物化学家很有价值,帮助他们从结构生物学角度理解突变导致功能丧失的原因,从而指导分子设计和靶点成药性评估。
在 AI 制药领域,能够通过 3D 结构变化来解释突变机理的模型,是真实研发场景中需要的实用工具。
📜Title: StructGuy: Data leakage free prediction of functional effects of genetic variants
🌐Paper: https://www.biorxiv.org/content/10.64898/2025.12.01.691563v1
2. 细胞 ABM 建模困局:告别僵化参数,拥抱「积木式」灵活性
从事计算辅助药物发现或系统生物学的同行,多半经历过这种挫败:试图用基于主体模型(Agent-Based Models, ABM)模拟药物在组织内的扩散或细胞反应,却发现手头的工具如同「水泥浇筑」。
Jonas Pleyer 的新文章精准击中了这一痛点。
被「写死」的生物学
现有 ABM 框架常将「细胞」定义为固定刚体,或预设特定网格环境。这如同想买乐高积木搞创作,厂家却只卖粘死的成品。
一旦研究对象涉及细胞变形或特殊微环境,就必须强行填入大量参数修补模型。模型因此臃肿不堪,参数估计(Parameter Estimation)沦为玄学,无法验证究竟是生物学机制在起作用,还是参数凑巧拟合。
我们需要「生物学的 PyTorch」
不妨参考数学工具。经典微分方程或深度神经网络(DNN)之所以强大,源于它们仅提供基础运算逻辑,而非预设解决方案。使用 PyTorch 搭建网络时,层数与连接方式完全由用户决定。
目前的 ABM 正缺乏这种底层灵活性。我们需要从零构建或大刀阔斧修改模型的能力,摆脱预定义几何形状或规则的束缚。
把细胞拆成积木
核心思路在于模块化(Building Block-based)。
直接模拟细胞的组成部分,将膜、细胞骨架、受体分布拆解为独立、可复用的数学组件。研究免疫细胞穿过血管壁时,调用「变形」和「粘附」组件;研究植物细胞时,加入「细胞壁」组件。
这种方法若能实现,我们将不再为新问题重复造轮子,而是建立通用且经验证的生物组件库。在药物研发中,这将允许针对特定靶点快速搭建高保真模拟环境,不再受困于标榜「通用」却难以精准模拟的商业软件。
📜Title: Agent-Based Modelling in Cellular Biology: Are we flexible yet?
🌐Paper: https://arxiv.org/abs/2511.12161v1
3. FoldSAE:用稀疏自编码器精准操控蛋白质折叠
RFdiffusion 是计算蛋白质设计领域的重磅工具,但其运作机制如同黑盒:输入提示词,输出结构,中间过程不透明。作者试图揭开这一黑盒,探究其内部齿轮的运转。
稀疏化是理解的关键
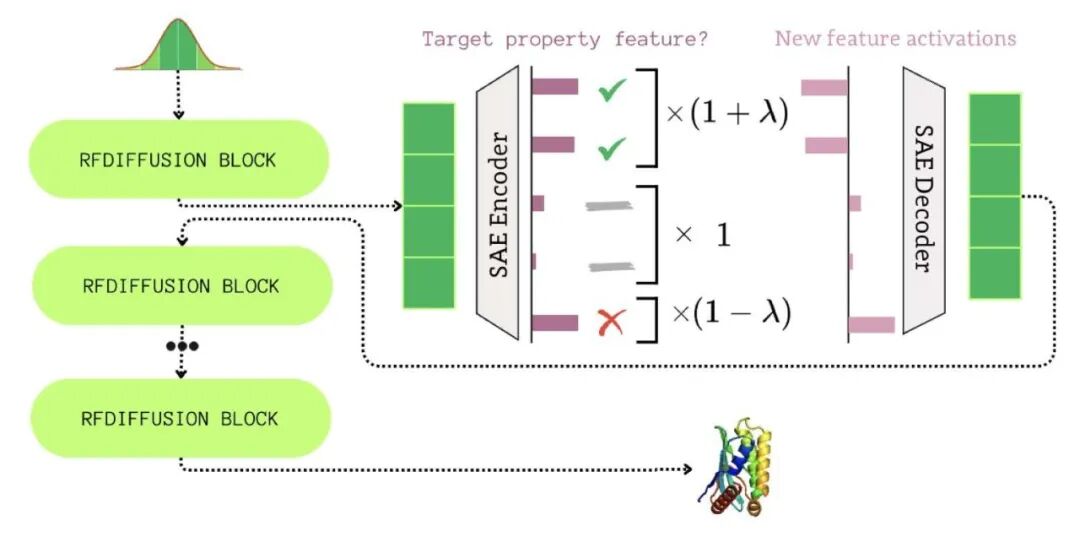
研究者提出 FoldSAE 框架,核心策略是利用稀疏自编码器(Sparse Autoencoders, SAE)处理 RFdiffusion 的内部数据。RFdiffusion 内部充斥着高度压缩、纠缠的稠密表征。作者利用 SAE 将这些稠密向量拆解为稀疏、可解释的特征,如同将混合果汁还原为原始成分,精准识别起关键作用的要素。
发现结构生成的「跷跷板 」
拆解后,研究者在潜空间(latent space)识别出具有「对抗性」的特征。这些特征如同跷跷板:增强某些特征会导致生成的蛋白质螺旋(Helix)结构增加,折叠片(Strand)结构减少。这种正负相关性揭示了蛋白质折叠过程中二级结构竞争的底层逻辑。
从「抽卡」到「工程设计 」
蛋白质设计因此摆脱了单纯的概率博弈。作者将这些特征转化为「控制旋钮」,通过放大或抑制特定特征,对生成的蛋白质骨架进行微调。例如,调节超参数即可增加 α-螺旋,在保持生物学合理性的前提下,精确控制二级结构含量。
未来的可能性
FoldSAE 推动了从「随机采样」向「精准工程」的转变。控制二级结构的逻辑未来可扩展至更复杂的属性,如溶剂可及性或配体结合位点的形成。计算辅助药物设计(CADD)正逐渐演变为理性的建筑设计。
📜Title: FoldSAE: Learning to Steer Protein Folding Through Sparse Representations
🌐Paper: https://arxiv.org/abs/2511.22519v1
4. AI+MD:攻克 EGFR 耐药突变新策略
EGFR 是小分子药物研发中著名的「不死反派」。从第一代吉非替尼到奥希替尼,癌细胞总能通过突变逃逸。本研究聚焦 L858R/T790M 双重突变,这是非小细胞肺癌 (NSCLC) 对一代和二代 TKI 产生耐药的主因。
突变的本质
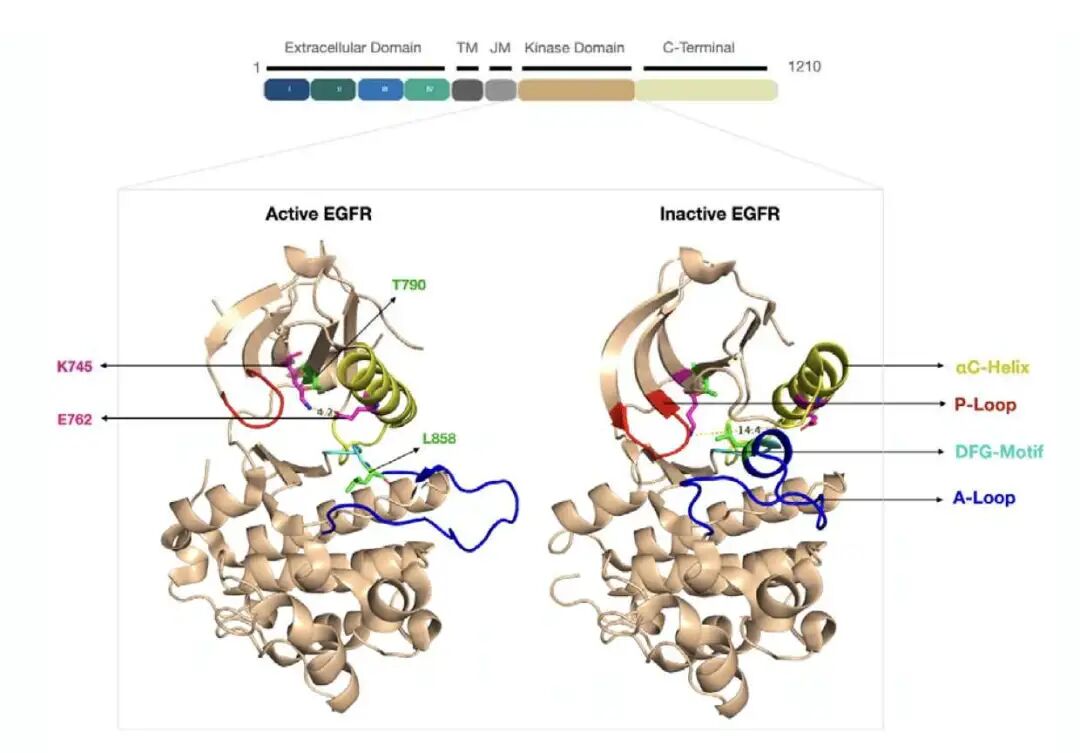
利用微秒级分子动力学 (MD) 模拟观察发现,L858R/T790M 突变改变了蛋白能量图景。即便没有配体,突变后的 EGFR 激酶也倾向于维持「活跃状态」 (active-like conformation)。
理想的变构抑制剂需如同一把强力「老虎钳」,将激酶强行扭回并锁死在「失活态」。
结合机制解析
分析已知变构抑制剂 EAI001 发现,其结合破坏了 K745 与 E762 间的盐桥,引发结构元件重排,增加了失活态构象的种群密度。仅依赖静态晶体结构不足以指导药物设计,必须通过 MD 模拟捕捉蛋白的动态变化。
AI 辅助筛选流程
研究团队设计了一套筛选组合拳,以寻找超越 EAI001 的分子:
- 初筛:进行大规模分子对接 (Docking)。
- 精选:利用机器学习打分函数 (SG-ML-PLAP) 重新排序,剔除高分但结合不稳定的假阳性分子。
- 验证:通过 MD 模拟和 MM/GBSA 计算,验证分子在动态环境下稳定 EGFR 失活构象的能力。
结果与前景
流程筛选出 10 个全新候选分子,计算预测效力优于 EAI001。尽管计算结果距离临床应用尚远,但这为克服 EGFR 耐药性提供了扎实的结构生物学切入点。针对变构位点的策略避开了拥挤的 ATP 结合口袋,有望为现有疗法无效的患者提供第四代药物研发线索。
📜Title: Design of Allosteric Inhibitors for Mutant EGFR by Combined use of Machine Learning and Molecular Dynamics Simulations
🌐Paper: https://www.biorxiv.org/content/10.64898/2025.12.01.691623v1
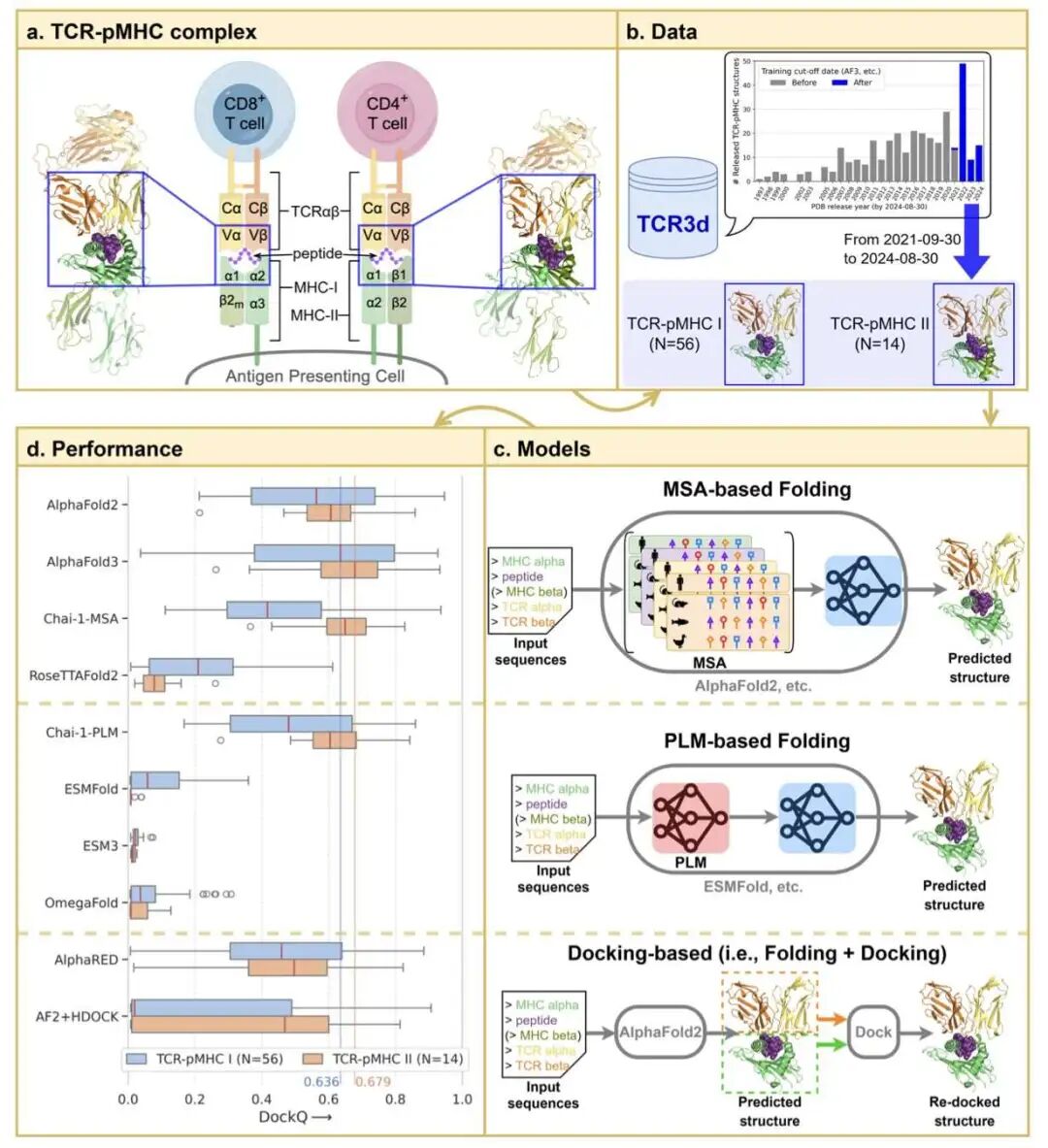
5. AF3 称霸 TCR-pMHC 预测,CDR3 分数成关键指标
TCR-pMHC(T 细胞受体 - 多肽 - 主要组织相容性复合体)的识别机制是免疫疗法与疫苗研发的基础。由于该三元复合物具有高柔性与多变性,准确模拟其结构极具挑战。BioRxiv 上发表的一项最新评测对比了主流结构预测工具,确认 AlphaFold3 (AF3) 为当前最优解。
AF3 的全面优势
研究选取了 70 个模型从未见过的 TCR-pMHC 复合物作为测试集。AlphaFold3 在与基于 MSA 的方法、PLM 及传统分子对接工具的对比中胜出。无论针对 Class I 还是 Class II 分子,它生成的结构在对接质量和原子精度上均表现最佳,有力提升了对免疫识别机制的理解。
CDR3 pLDDT:结合质量的代理指标
通常 pLDDT 仅代表模型对预测结构的置信度。但在 TCR-pMHC 场景下,CDR3 区域的 pLDDT 分数直接关联预测的结合质量。
若模型对 CDR3 这一关键识别环的结构评分较高,整体复合物的对接通常可靠。该分数还能敏锐捕捉单点突变引起的亲和力变化。在药物设计中,仅凭此分数即可初步筛选增强或破坏结合的突变,减少了对昂贵湿实验的依赖。
速度与精度的平衡
工业界应用需兼顾速度与精度。测试显示,采用加速版 MSA 策略可大幅降低计算时间且不损耗预测精度。计算机模拟大规模 TCR-pMHC 库筛选已具备现实操作性。
该研究确立了 AF3 的优势,并提供了一套实用评估标准。观察 AI 预测的 TCR 结构时,CDR3 的 pLDDT 分数所蕴含的信息值得重点关注。
📜Title: Benchmarking TCR-pMHC structure prediction: A unified evaluation and CDR3-based functional insights
🌐Paper: https://www.biorxiv.org/content/10.64898/2025.11.30.691400v1