使用深度学习方法预测噬菌体特异性蛋白质完整代码实现,含数据集。 该方法对噬菌体特异性蛋白(TerL、Portal和TerS)具有良好的预测精度,也可用于从病毒组数据预测序列。 CNN可以自动学习蛋白质序列模式,同时根据学习到的氨基酸序列模式构建预测模型。 与比对方法相比,CNN是基于氨基酸的模型的自然泛化,可以发现高纬度地区病毒蛋白质的相似性。 在病毒蛋白序列预测问题上,更灵活的 CNN 模型确实优于比对方法。
在生物学领域,对噬菌体特异性蛋白质的预测一直是个关键课题。今天咱就来唠唠如何用深度学习方法实现对噬菌体特异性蛋白质(TerL、Portal 和 TerS)的预测,不仅有完整代码,还包含数据集相关内容。
为什么选择深度学习方法
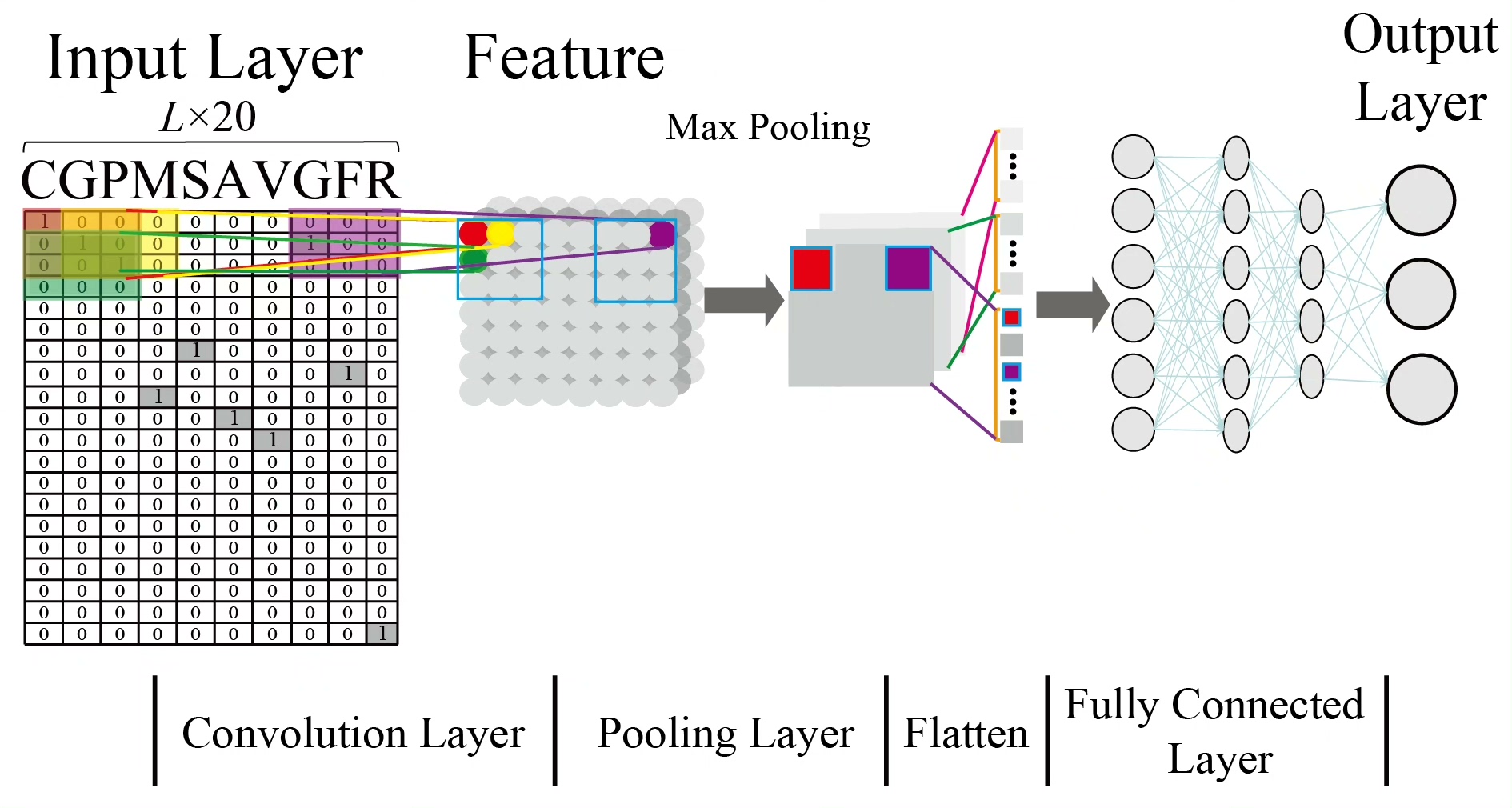
传统的比对方法在处理病毒蛋白序列预测问题时,灵活性欠佳。而 CNN(卷积神经网络)就不一样了,它可以自动学习蛋白质序列模式 ,并依据学习到的氨基酸序列模式构建预测模型。这就好比 CNN 有一双"慧眼",能自动识别蛋白质序列里那些微妙的特征。与比对方法相比,CNN 基于氨基酸的模型能自然泛化,在发现高纬度地区病毒蛋白质的相似性上,有着独特的优势。在实际应用中,更灵活的 CNN 模型确实在病毒蛋白序列预测问题上优于比对方法 。
数据集
首先,数据集是整个预测任务的基石。对于噬菌体特异性蛋白质预测,我们需要收集大量已知的噬菌体特异性蛋白(TerL、Portal 和 TerS)序列数据。这些数据可以从公开的生物数据库,如 NCBI 等获取。假设我们已经下载好了数据集,它可能是一个包含蛋白质序列及其对应标签(是否为目标噬菌体特异性蛋白)的文件,格式可能是 FASTA 或者 CSV 等。例如,以下是一个简化的 CSV 数据集示例:
text
sequence,label
MKWVTFISLLFLFSSAYS,1
AGKLVFGGFGFGDAA,0这里第一列是蛋白质序列,第二列是标签,1 代表是目标噬菌体特异性蛋白,0 代表不是。
代码实现
下面就是激动人心的代码部分啦,咱们使用 Python 和 PyTorch 框架来实现基于 CNN 的噬菌体特异性蛋白质预测。
数据预处理
python
import pandas as pd
from torch.utils.data import Dataset, DataLoader
from sklearn.preprocessing import LabelEncoder
class ProteinDataset(Dataset):
def __init__(self, csv_file):
data = pd.read_csv(csv_file)
self.sequences = data['sequence'].tolist()
self.labels = data['label'].tolist()
self.encoder = LabelEncoder()
self.labels = self.encoder.fit_transform(self.labels)
def __len__(self):
return len(self.sequences)
def __getitem__(self, idx):
sequence = self.sequences[idx]
label = self.labels[idx]
return sequence, label
csv_file = 'your_dataset.csv'
dataset = ProteinDataset(csv_file)
dataloader = DataLoader(dataset, batch_size=32, shuffle=True)代码分析:
- 我们定义了一个
ProteinDataset类,它继承自torch.utils.data.Dataset。这个类负责读取 CSV 数据集文件。 - init 方法中,我们读取文件,提取蛋白质序列和标签。并用
LabelEncoder对标签进行编码,使其变成模型能处理的数值形式。 - len 方法返回数据集的长度,也就是序列的数量。
- getitem 方法根据索引返回对应的序列和标签。最后通过
DataLoader对数据集进行包装,设置批量大小为 32 并打乱数据顺序。
构建 CNN 模型
python
import torch
import torch.nn as nn
class ProteinCNN(nn.Module):
def __init__(self):
super(ProteinCNN, self).__init__()
self.conv1 = nn.Conv1d(in_channels=20, out_channels=32, kernel_size=3)
self.relu1 = nn.ReLU()
self.pool1 = nn.MaxPool1d(kernel_size=2)
self.fc1 = nn.Linear(32 * (len(max(dataset.sequences, key=len)) - 2) // 2, 64)
self.relu2 = nn.ReLU()
self.fc2 = nn.Linear(64, 2)
def forward(self, x):
x = self.conv1(x)
x = self.relu1(x)
x = self.pool1(x)
x = x.view(-1, 32 * (len(max(dataset.sequences, key=len)) - 2) // 2)
x = self.fc1(x)
x = self.relu2(x)
x = self.fc2(x)
return x
model = ProteinCNN()代码分析:
- 定义
ProteinCNN类,继承自nn.Module。这是我们构建的 CNN 模型主体。 - 在 init 方法中,我们定义了一个一维卷积层
conv1,输入通道数设为 20(因为氨基酸种类有 20 种),输出通道数为 32,卷积核大小为 3。接着是激活函数ReLU和最大池化层pool1。 - 然后是两个全连接层
fc1和fc2。在计算fc1的输入维度时,要考虑卷积和池化后的特征图大小。 forward方法定义了数据在模型中的前向传播路径,数据依次经过卷积、激活、池化和全连接层。
训练模型
python
import torch.optim as optim
criterion = nn.CrossEntropyLoss()
optimizer = optim.Adam(model.parameters(), lr=0.001)
num_epochs = 10
for epoch in range(num_epochs):
running_loss = 0.0
for i, data in enumerate(dataloader, 0):
sequences, labels = data
optimizer.zero_grad()
outputs = model(sequences)
loss = criterion(outputs, labels)
loss.backward()
optimizer.step()
running_loss += loss.item()
print(f'Epoch {epoch + 1}, Loss: {running_loss / len(dataloader)}')代码分析:
- 我们定义了损失函数
CrossEntropyLoss,它结合了Softmax激活函数和NLLLoss,很适合多分类问题。 - 使用
Adam优化器来更新模型参数,学习率设为 0.001。 - 在训练循环中,我们遍历数据集的每个批次,每次迭代都将梯度清零,前向传播得到输出,计算损失,反向传播计算梯度,最后更新模型参数。每一个 epoch 结束后,打印当前 epoch 的平均损失。
通过以上的数据集准备和代码实现,我们就完成了基于深度学习 CNN 方法的噬菌体特异性蛋白质预测。这种方法在对噬菌体特异性蛋白(TerL、Portal 和 TerS)的预测上有着良好的精度,并且也可用于从病毒组数据预测序列,为生物学研究提供了有力的工具。