在单细胞测序数据分析中,barcodes、features和matrix是三个最核心的基础文件,它们共同构成了所有分析的基石。
| 特性维度 | 细胞条形码 (barcodes,BC) | 表达矩阵 (Matrix) |
|---|---|---|
| 核心角色 | 细胞身份证 | 核心数据账本 |
| 功能定位 | 定位数据属于哪个细胞 | 分析每个细胞中基因表达量有多少 |
| 数据结构 | 表达矩阵的列名 (Column Names) | 一个二维表格:行是基因,列是细胞,元素是表达量 |
| 文件表现 | 通常为 barcodes.tsv文件,每行是一个条形码序列 | 通常为 matrix.mtx文件(稀疏矩阵格式),存储非零表达值 |
| 依赖关系 | 表达矩阵的列数由有效的细胞条形码数量决定 | 表达矩阵的列标识完全依赖于细胞条形码文件 |
一、进入GEO官网:https://www.ncbi.nlm.nih.gov/geo/
在GEO(Gene Expression Omnibus)数据库中下载单细胞数据时,最常见的数据存储和提供形式主要有以下四种类型:
- 10x Genomics 标准格式(最主流)
在GEO的数据集中,我们通常会找到一个包含以下三个核心文件的文件夹(通常以 *_filtered_feature_bc_matrix.tar.gz或类似名称的压缩包提供):
barcodes.tsv(.gz): 包含所有检测到的细胞条形码(Cell Barcode),每个条形码代表一个可能的细胞。
features.tsv(.gz) 或 genes.tsv(.gz): 包含所有被检测的基因(或转录本、抗体标签)的标识符和名称。
matrix.mtx(.gz): 以稀疏矩阵格式存储的基因表达计数矩阵,行对应基因,列对应细胞。
- H5 格式(高效二进制格式)
这是一种用于存储大规模数据的二进制文件格式。在单细胞上下文中,它通常是将上述10x标准格式的三个文件整合到了一个单一的 .h5文件中。
典型命名: *_filtered_feature_bc_matrix.h5
优势: 文件单一,便于管理和传输,存储效率高。
- R 数据文件(可直接使用的分析对象)
部分数据提交者会将已初步处理好的单细胞数据直接保存为R语言特有的数据文件格式,供其他研究者快速载入。
.rds文件: 存储单个R对象(如一个Seurat对象或表达矩阵)。使用 readRDS()函数读取。
.RData或 .rda文件: 可存储一个或多个R对象。使用 load()函数读取,对象会被载入当前工作环境。
优势: 读取速度极快,且能保留完整的对象结构和中间分析结果。
- 压缩文本矩阵(通用表格格式)
这是一种比较传统和通用的数据提供方式,将表达矩阵保存为纯文本表格,并进行压缩以减小体积。
格式: 通常是 .txt.gz或 .csv.gz文件。
内容: 行是基因,列是细胞(或样本),表格内的值为表达量。有时也可能提供转置后的格式(行是细胞,列是基因)。
如何读取: 可以使用 data.table::fread()、read.delim()等函数读取,或在Excel中打开(不推荐用于大型数据)。读取后通常需要转换为矩阵或数据框,才能用于创建Seurat对象。
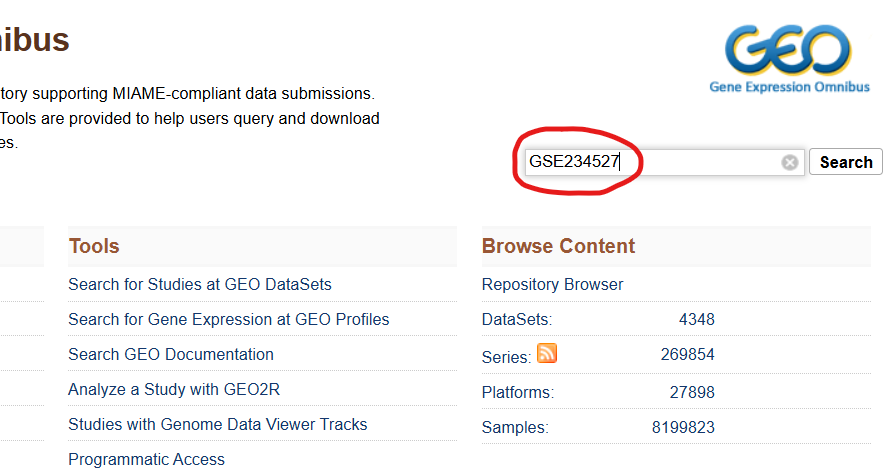
二、以"GSE234527"为例子:读取一个10× Genomics格式文件
1、搜索"GSE234527":
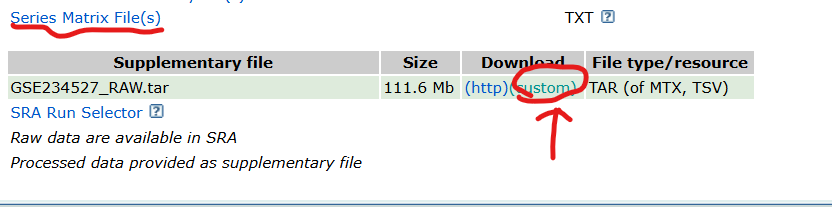
2、在补充材料里下载(点击"custom")

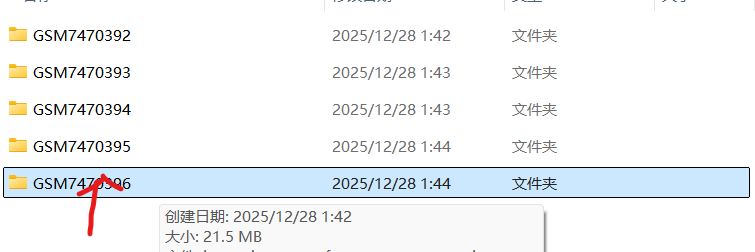
3、在桌面解压缩,分类整理:数据来自五个不同样本,分成五类,每个样本包含多个细胞
修改前:

修改后:
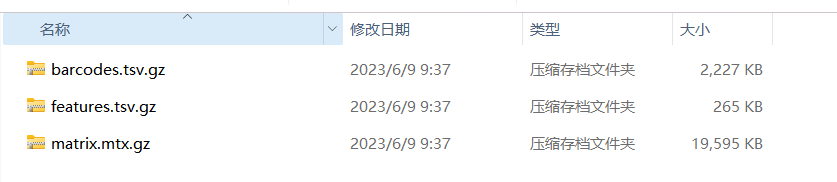
*修改的目的是为了更好地用R语言进行数据处理
详细过程请参考:https://www.bilibili.com/video/BV1Ct421j7X3?spm_id_from=333.788.videopod.sections&p=4
(B站:生信幻想家------单细胞数据分析)