本文内容速览:
此前,小远给大家介绍了ATAC-seq与3C(染色质构象捕获技术)的基本知识,相关推文"解析表观遗传学的工具------ATAC-seq(一)","解析表观遗传学的工具------ATAC-seq(二)","植物学家们的美好愿景------建立植物ENCODE(pENCODE)","如何研究"暗物质"------增强子?(一)"。ATAC-seq能够在全基因组范围内精确检测染色质的开放程度,通过分析不同条件下染色质开放程度的动态变化,我们可以获取大量与基因表达调控相关的关键信息。而染色质构象捕获技术及其衍生技术,则主要用于解析基因组中不同区域之间的染色质空间互作关系。今天小远将给大家介绍的Hi-C技术,正是染色质构象捕获技术的延伸,是表观遗传学研究中的一项重要研究工具。该技术能够高通量地捕获全基因组范围内的染色质三维交互信息,揭示染色质在空间结构上的组织模式。结合ATAC-seq所反映的染色质可及性状态,Hi-C技术能够帮助我们更全面地理解染色体结构变化与表观遗传调控之间的内在关联,共同揭示基因表达调控在三维空间层面的复杂机制。
0 1
Hi-C技术的发展历程
2002年,Job Dekker课题组在Science杂志上发表了一篇题为"Capturing chromosome conformation"的里程碑式论文,首次开发了基于DNA片段邻近连接的3C技术(Chromosome conformation capture,3C)。该技术利用甲醛交联和限制性内切酶处理,将空间上相邻的染色质片段连接,再通过定量PCR对特定位点间的互作进行定量分析,实现了对染色质"one-vs-one"的靶向互作检测 (Dekker et al., 2002)。然而,3C技术靶向性强的特点也限制了其通量。为突破 3C 技术的检测瓶颈,基于其核心原理开发了系列衍生技术:环状染色质构象捕获技术(Circular chromatin conformation capture,4C)通过环状连接与反向PCR扩增策略,实现了从单个已知位点出发,无偏筛选全基因组范围内所有互作伙伴的"one-vs-all"检测 (Zhao et al., 2006);而染色质构象捕获碳拷贝技术(Chromatin conformation capture carbon copy,5C)则借助多重引物扩增与连接介导的测序文库构建,实现了特定基因组区域内"many-vs-many"的高通量互作检测 (Dostie et al., 2006)。尽管4C和5C技术显著扩展了检测范围,但仍未摆脱 "靶向区域或单个位点" 的限制,无法实现全基因组尺度、无偏倚的染色质互作全景解析。
这一技术瓶颈直至 2009 年被打破,Lieberman-Aiden课题组在Science杂志上发表了一篇题为"Comprehensive mapping of long-range interactions reveals folding principles of the human genome"的研究论文。该研究将3C技术的"邻近连接"核心思路与高通量测序技术深度融合,开发出全基因组染色质构象捕获技术(Hi-C),首次实现了"all-vs-all"的染色质互作图谱绘制,无偏倚地揭示了全基因组范围内的染色质三维互作网络。更重要的是,该研究通过Hi-C数据首次提出染色体"分区/分层"的核心概念,为理解基因组空间组织规律奠定了理论基础 (Lieberman-Aiden et al., 2009)。
自此以后,Hi-C技术不断在概念、分辨率、效率和数据分析方法上改进与完善。它不仅能够精准绘制染色质拓扑关联结构域(TADs),还能鉴定大量调控基因表达的染色质环(如增强子-启动子环),从而极大地推动了三维基因组学研究的纵深发展。
表1 3C、4C、5C及Hi-C技术的区别(Hakim and Misteli, 2012)。
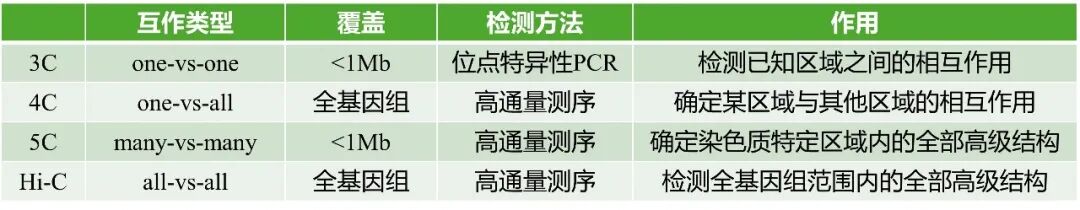
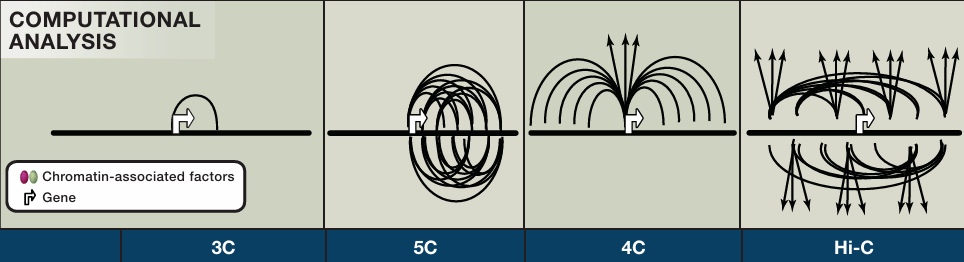
图1 3C、4C、5C及Hi-C技术原理区别(Hakim and Misteli, 2012)。
0 2
Hi-C技术原理及流程
染色质的三维结构是基因组在细胞核内实现精准调控的核心空间基础,其动态组织方式直接关联基因表达调控、细胞分化、发育稳态等关键生物学过程,甚至在疾病发生(如肿瘤)和植物-微生物互作等场景中扮演着决定性角色(Deng et al., 2022)。不同于线性的DNA序列,染色质三维结构本质是基因组与组蛋白、转录因子、染色质重塑复合物等蛋白质组装形成的动态空间构象,从宏观到微观,染色质的三维结构呈现出清晰的层级化组织特征(图1):染色体区域分别包含转录活跃状态的常染色质与惰性状态异染色质的A/B区室,作为结构和功能边界的拓扑结合结构域,以及实现远端调控元件(如增强子、沉默子)与启动子特异性结合的染色质环(Liu et al., 2024)。深入解析染色质三维结构的层级组织规律与动态变化,有助于揭示"基因组序列---空间构象---基因活性---细胞功能"的内在关联,使得调控信号能够精确传递。
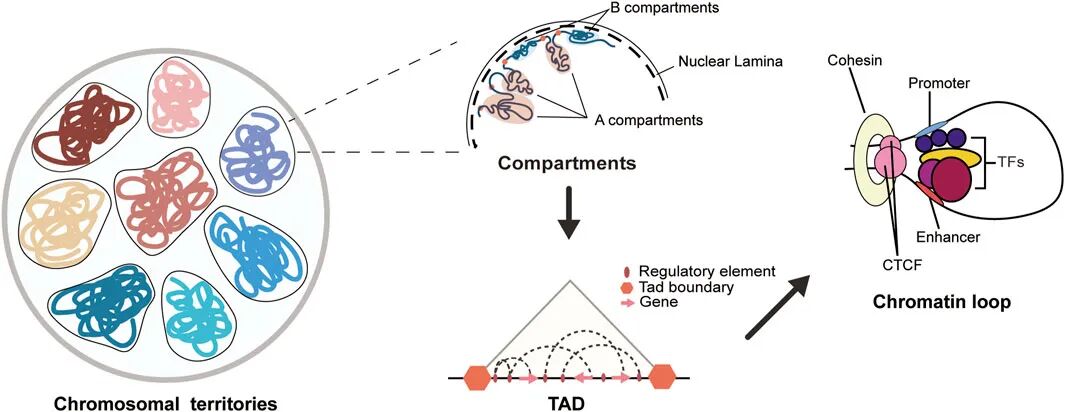
图2 染色质三维结构图 (Liu et al., 2024)。细胞核中基因组的三维结构根据层级依次为染色体区域、A/B区室、TAD和染色质环。
Hi-C技术是在3C基础上发展起来的高通量染色质构象捕获技术,它通过与高通量测序相结合,实现了对全基因组范围内染色质相互作用无偏倚的系统性检测 (Lieberman-Aiden et al., 2009; Belton et al., 2012)。其核心实验流程旨在捕获并锁定染色质在空间上的近距离接触,主要包含以下几个关键步骤(图2):
(1)交联与固定:首先使用甲醛处理细胞,使空间上相互靠近的染色质片段与蛋白质之间发生共价交联,从而固定它们在三维空间中的相互作用。
(2)酶切与标记:利用限制性内切酶将交联的基因组DNA进行切割,产生带有粘性末端或平末端的片段。随后,将这些末端进行补平修复,并在这一过程中掺入被生物素标记的dNTP,对潜在的相互作用位点进行特异性标记。
(3)连接与捕获:在稀释条件下使用DNA连接酶进行片段间的连接。由于交联使得空间邻近的片段被拉拢在一起,连接反应主要发生在原本在三维空间中紧密接触的染色质片段之间,形成嵌合DNA分子。
(4)纯化与建库:解交联,纯化得到DNA,并将其随机打断为更小的片段。利用链霉亲和素磁珠特异性地富集内部带有生物素标记的片段(即连接点片段),从而构建出用于高通量测序的文库。
(5)测序与分析:对构建好的文库进行高通量测序,产生的海量配对序列读数再经生物信息学流程进行处理与比对,最终转化为全基因组水平的染色质交互矩阵,揭示染色质的空间组织模式。
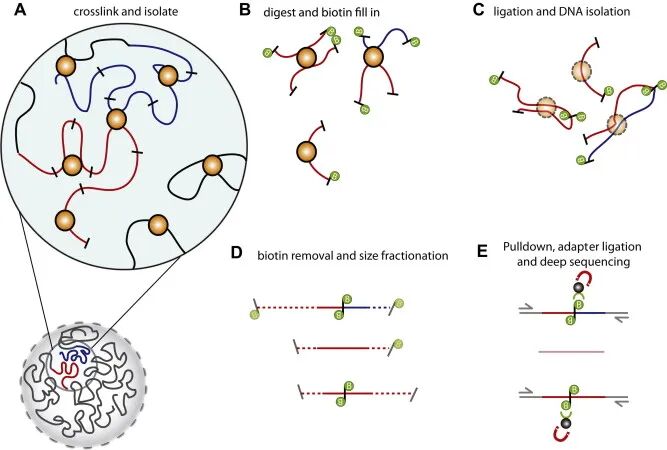
图3 Hi-C技术流程 (Belton et al., 2012)。(A)Hi-C通过用甲醛共价交联蛋白质/DNA复合物来检测染色质在染色体内部和染色体之间的相互作用;(B)染色质被限制性内切酶切割,产生的末端用生物素标记;(C)交联复合体中的DNA被连接形成嵌合DNA分子;(D)从DNA线性片段末端去除生物素,DNA分子被断裂以缩小整体大小;(E)含有内部生物素标记的DNA片段被链霉亲和素磁珠拉下以进行深度测序。
0 3
Hi-C技术在植物中的应用
Hi-C技术作为染色质三维结构解析领域的革命性突破,为探索真核基因组的空间组织规律开辟了全新路径。与动物研究相比,Hi-C在植物领域的应用仍处于发展阶段,一方面源于植物细胞特有的细胞壁结构、复杂的基因组重复序列及组织特异性染色质特征带来的技术挑战,另一方面也与植物三维基因组功能研究的起步较晚相关。即便如此,Hi-C技术仍在植物染色质三维结构解析中发挥了不可替代的作用,为解析植物基因组的空间组织与基因表达调控、环境胁迫响应、生长发育调控等过程的关联提供了关键支撑。
3.1
植物基因组组装和染色体拼接
Hi-C技术在植物基因组学中的最重要应用之一是辅助完成染色体级基因组组装。由于植物基因组普遍具有规模大、重复序列比例高、结构变异丰富等特点,常规的短读长或长读长组装方法在contig锚定与染色体排序方面面临巨大困难。Hi-C所提供的远距离染色质互作信息,可根据物理邻近性将序列片段准确定位到染色体上,从而提高组装完整性与准确度,这一特性使Hi-C成为构建高质量植物基因组不可或缺的关键技术。
2021年4月,中国科学院分子植物科学卓越创新中心韩斌课题组与中科院植物研究所北方资源植物重点实验室桑涛课题组合作在Nature Communications 杂志上发表了一篇题为"Chromosome-scale assembly and analysis of biomass crop Miscanthus lutarioriparius genome"的研究论文,该研究以多年生禾本科芒属南荻为研究对象,对其基因组进行测序和绘制(图4)。南荻基因组较大且重复序列含量高,其基因组一直被视为较难测序组装的复杂植物基因组。通过联合Oxford Nanopore测序和Hi-C技术,该研究报道了一个染色体级别组装的南荻基因组,大小2.07Gb,覆盖全基因组的96.64%,contig N50为1.71Mb。着丝粒和端粒序列分别组装至19条染色体和10号染色体,并且利用着丝粒卫星重复序列证实南荻的异源四倍体起源 (Miao et al., 2021)。
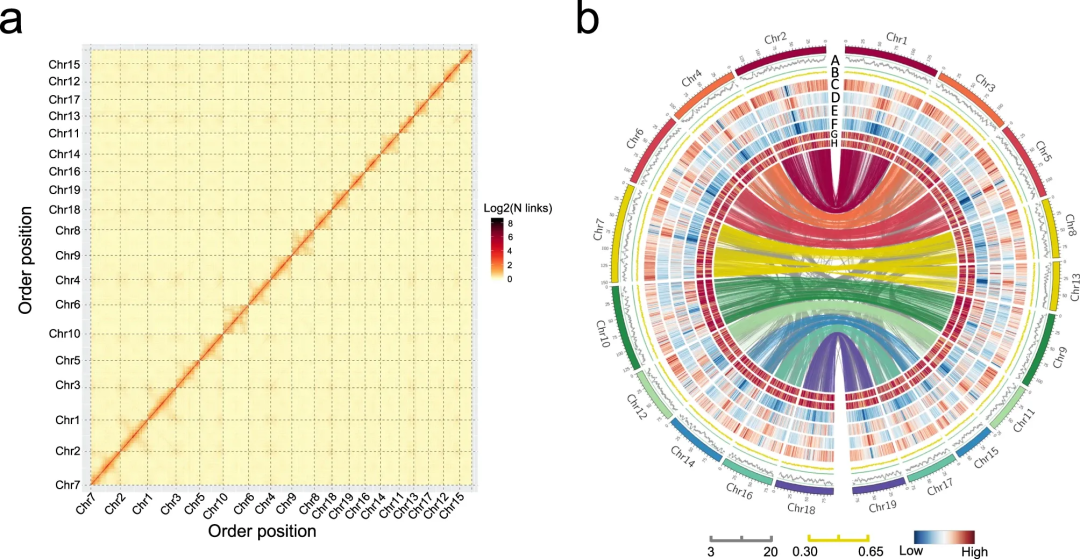
图4 南荻基因组组装概述 (Miao et al., 2021)。(a)南荻全基因组Hi-C图谱。聚类后热图显示了LACHESIS流程生成的重叠群之间的Hi-C互作密度;(b)南荻基因组组装的Circos图。最外层彩色环为19条假染色体(刻度:5Mb/格)。各轨道依次表示:LAI得分(A)、基于100kb非重叠窗口计算的GC含量(B)、基因(C)、Gypsy(D)、Copia(E)、DNA转座子(F)密度,以及根茎(G)和侧芽(H)的转录组表达水平。最内层展示了染色体间的共线性关系:其中上层的彩色链接代表由近期南荻全基因组复制事件(WGD)产生的共线性区块,下层的灰色链接则代表由更早复制事件产生的同源基因对。
3.2
功能性互作识别
Hi-C能够识别染色体中远程的调控互作,如增强子-启动子互作、调控环状结构以及条件特异性的长程调控关系。植物基因组中广泛存在分散型调控元件,这类互作对于调控响应、发育和表型形成具有重要意义。
2024年11月,北京大学现代农学院周岳课题组在Genome Biology杂志上发表了一篇题为"Promoter capture Hi-C identifies promoter-related loops and fountain structures in Arabidopsis"的研究论文,该研究通过启动子捕获Hi-C(PCHi-C)技术鉴定出与有序染色质结构相关的启动子相关环,并揭示了"喷泉结构"形成和维持的分子机制 (Wang et al., 2024)。
PCHi-C作为Hi-C技术的重要延伸,能高效捕获启动子与基因组其他区域的相互作用。相比常规Hi-C,该技术将启动子相关相互作用的检测灵敏度提升了近十倍,从而能够系统识别启动子相关染色质环。基于此,作者通过统计模型校正距离效应后,从全基因组互作数据中鉴定出27,946个显著的启动子相关染色质环。进一步整合ATAC-seq数据,将启动子相互作用区域划分为基因体、基因间区与近端启动子区域(图5)。基序富集分析显示,C2C2dof、MYB121及PCF转录因子结合的顺式调控元件在以上三类区域中均显著富集,提示这些因子可能共同参与介导启动子相关染色质环的形成。
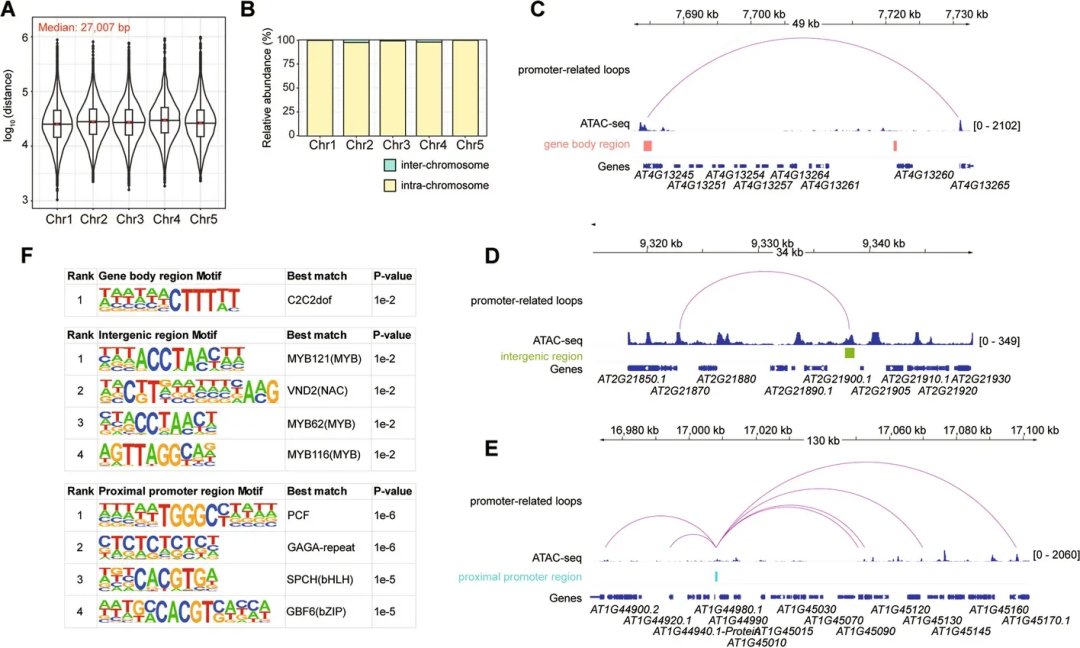
图5 启动子与基因体、基因间和近端启动子区域发生相互作用 (Wang et al., 2024)。(A) 小提琴图及对应箱型图展示了各染色体中启动子相关染色质环的长度分布,面板左上角标注了启动子相关染色质环的中位长度;(B)条形图显示了各染色体内部与染色体间启动子相关染色质环的相对丰度; (C)、(D)和(E)分别展示了基因体环(C)、基因间区环(D)和近端启动子环(E)的分布情况,其中红色、绿色和蓝色方框分别代表基因体、基因间及近端启动子区域;(F)显示了在基因体(顶部)、基因间(中部)和近端启动子区域(底部)显著富集的基序,并标注了识别前四位重要基序的转录因子及其对应P值。
作者进一步分析了启动子相关染色质环的分布模式,发现它们主要与高阶染色质结构------如拓扑关联域(TADs)和此前在拟南芥中未被报道的"喷泉结构"存在关联(图6)。研究基于Hi-C数据在拟南芥中鉴定出1361个喷泉结构,该结构在动物中已有报道,但在拟南芥中是首次发现。分析显示,启动子相关染色质环的相互作用程度与基因转录水平呈负相关,提示这些环可能参与转录抑制。与此同时,喷泉结构的强度与其中包含的环数量呈正相关,表明启动子相关染色质环对染色质结构的维持具有调控作用。进一步研究发现,喷泉中心两侧的环锚点区域显著富集H3K4me3修饰,且这些锚点之间存在跨越喷泉中心的互作。为探究其分子机制,作者聚焦于拟南芥主要的H3K4三甲基转移酶ATXR3。通过整合Hi-C、PCHi-C及RNA-Seq数据,研究证实ATXR3对维持喷泉内部的染色质环至关重要,并在此过程中发挥抑制喷泉相关基因转录的关键功能。
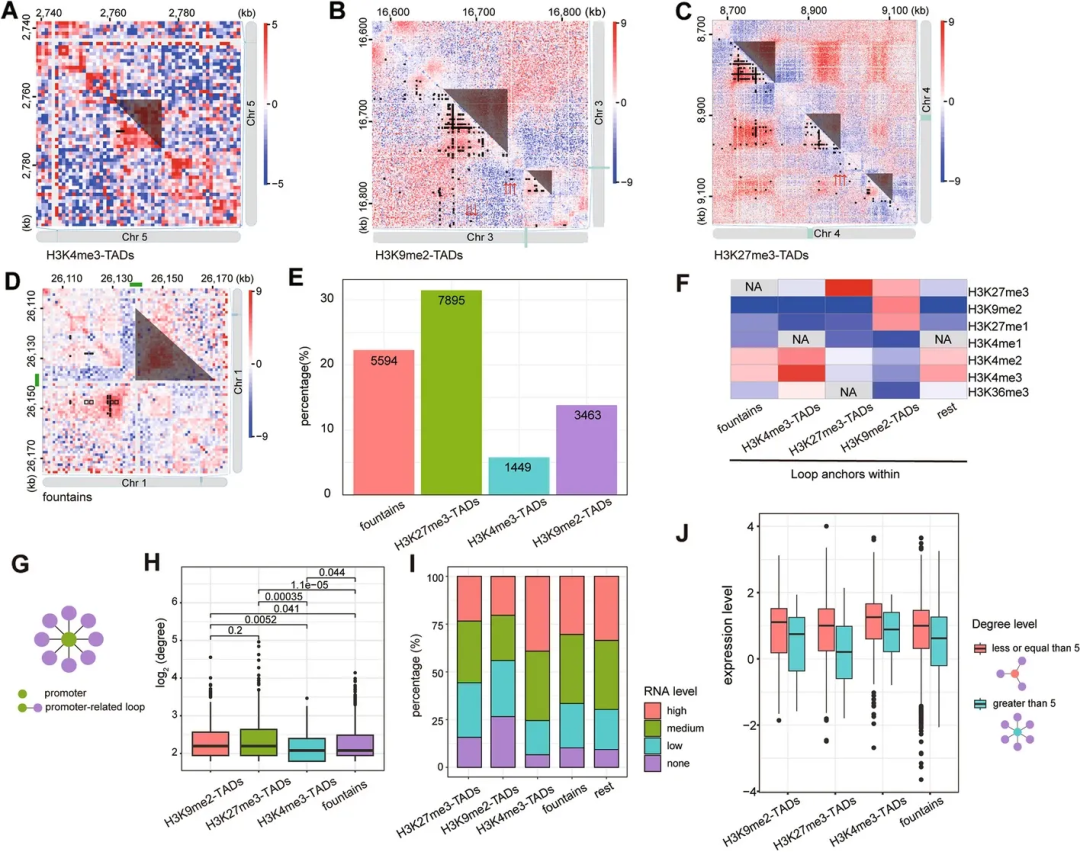
图6 启动子相关染色质环主要与不同的拓扑关联结构域(TAD)及喷泉结构相关,其功能在于抑制基因转录 (Wang et al., 2024)。(A)、(B)和(C)分别展示了H3K4me3-、H3K9me2-和H3K27me3-TAD区域的接触热图;(D)在O/E互作热图中展示了一个典型的喷泉结构;(E)展示了与每种高级染色质结构相关的启动子相关染色质环的比例及数量;(F)热图展示了与H3K9me2-TAD、H3K27me3-TAD、H3K4me3-TAD、喷泉结构或其他环相关的启动子相关染色质环锚定基因的相对组蛋白修饰水平;(G)启动子互作程度示意图。绿色和紫色圆圈分别代表启动子及其互作区域,圆圈间的连线代表启动子相关染色质环:(H)箱型图展示了与H3K9me2-TAD、H3K27me3-TAD、H3K4me3-TAD及喷泉结构相关的各启动子相关染色质环锚定基因的互作程度;(I)条形图显示了与H3K9me2-TAD、H3K27me3-TAD、H3K4me3-TAD及喷泉结构相关的启动子相关染色质环锚定基因的比例表达水平;(J)箱型图展示了与上述结构相关的启动子相关染色质环锚定基因的表达水平。
3.3
胁迫与发育下植物3D基因组的动态结构
植物的不同组织和发育阶段往往具有明显不同的三维染色体构象,且在面对高温、低温、干旱、盐胁迫等环境条件时,其三维染色体结构会迅速而深刻地发生重组。Hi-C技术可以捕获这些动态的空间结构变化,包括区室转换、长程互作的增强或削弱、染色体折叠方式的调整等,从而揭示胁迫响应基因的调控机制。
2021年3月,中国农业科学院生物技术研究所谷晓峰课题组在BMC Biology杂志上发表了一篇题为"Reorganization of the 3D chromatin architecture of rice genomes during heat stress"的研究论文,首次揭示了籼稻和粳稻染色质三维空间结构在高温胁迫下发生重组的动态变化(图7)。
该研究利用Hi-C、ATAC-seq和RNA-seq等技术,系统探究了粳稻品种日本晴与籼稻品种93-11在高温胁迫下染色质三维结构及其可及性的动态变化。结果表明,高温胁迫显著影响两个品种的染色质空间构象,引发多层级结构重组,包括A/B区室转换、拓扑关联结构域尺寸增大以及短程互作减弱。进一步分析发现,上述三维结构变化与染色质可及性(以ATAC-seq测序信号富集分布为指标)的重新分布密切相关。值得注意的是,籼稻品种93-11在高温响应中表现出更为显著的基因表达动态调整和染色质可及性变化,这一特征与其较强的耐热表型相一致 (Liang et al., 2021)。
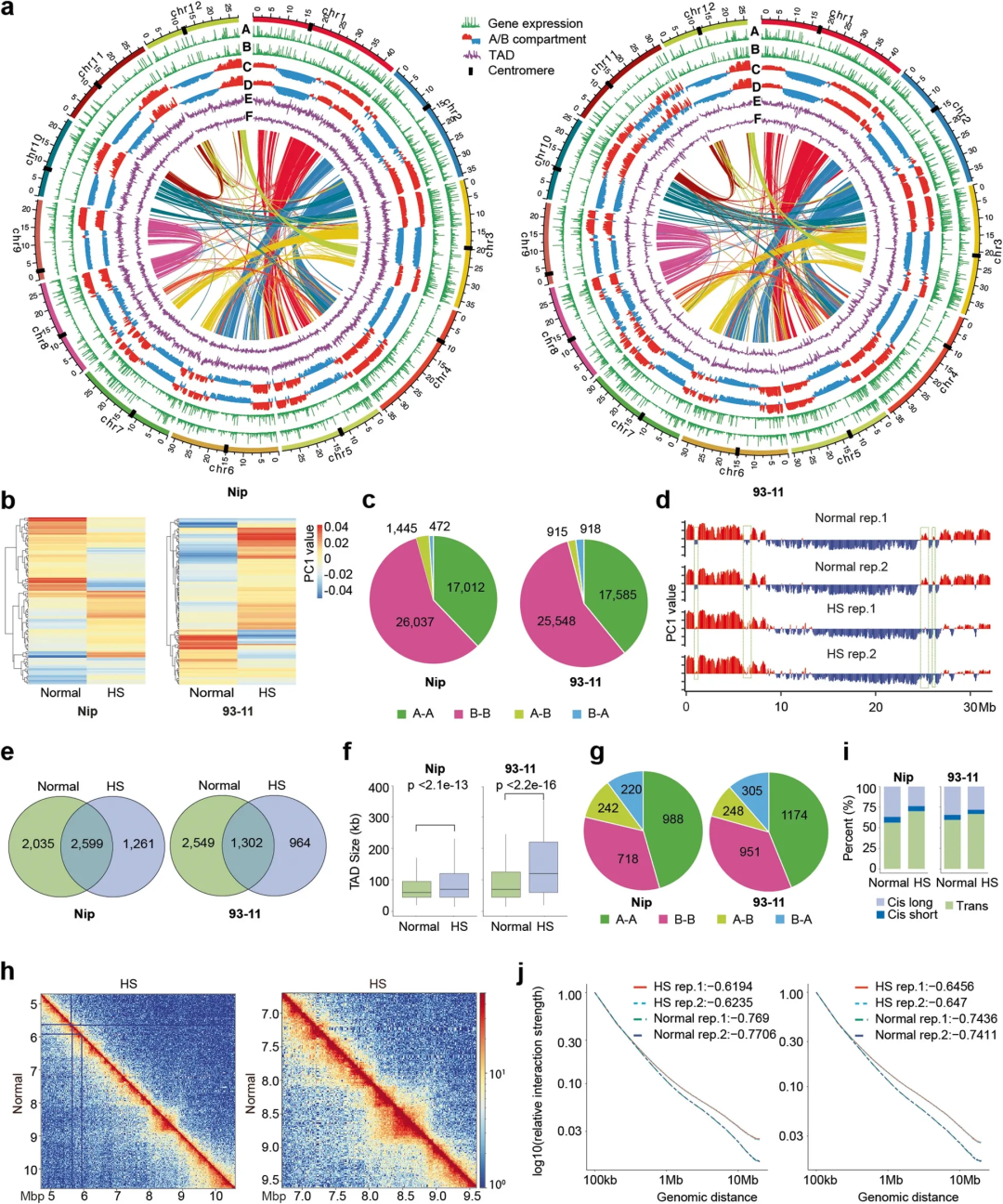
图7 水稻日本晴与93-11染色质三维结构响应热胁迫的动态变化 (Liang et al., 2021)。(a)日本晴(左图)与93-11(右图)基因组染色质三维结构的环形图;(b)日本晴与93-11中由热胁迫诱导发生区室转换区域的PC1分数热图;(c)在热胁迫下,日本晴与93-11全基因组区室数量按稳定区室(A-A和B-B)与转换区室(A-B和B-A)的划分;(d)93-11品种6号染色体的PC1分数,绿色框突出显示了在热胁迫下从A转换为B以及从B转换为A的区域;(e)维恩图分别显示日本晴与93-11中共有及样品特异性的TAD数量;(f)箱线图比较了日本晴与93-11在正常与热胁迫条件下TAD的大小;(g)在热胁迫下丢失TAD边界的区域中,饼图分别展示了日本晴与93-11局部区室按稳定型(A-A与B-B)及转换型(A-B与B-A)划分的数量分布;(h)日本晴正常与热胁迫样品中11号染色体某一区域(5-kb分辨率)Hi-C互作矩阵的比对示例;(i)条形图比较日本晴与93-11在正常与热胁迫条件下不同类型相互作用所占的百分比。根据距离,互作分为顺式长程(> 20kb)、顺式短程(< 20kb)和反式互作;(j)在热胁迫下,日本晴与93-11互作强度随距离变化的曲线图。
小远叨叨
小远今天简要介绍了Hi-C技术的原理、发展背景及应用实例。通过相关文献可以看出,Hi-C技术仍在持续发展,尤其在植物染色体三维结构解析领域具有广阔的发展潜力。该技术常与基因组学、转录组学及其它表观组学技术相结合,从而系统阐释染色体空间构象与功能的关联。小远最后补充一下,伯远生物可提供Hi-C、ATAC-seq、BS-seq、RNA-seq等多项组学技术服务,致力于为您的科学研究提供支持,助力发表高水平学术成果!
References:
BELTON J M, MCCORD R P, GIBCUS J H, et al. 2012. Hi-C: a comprehensive technique to capture the conformation of genomesJ. Methods, 58: 268-276.
DEKKER J, RIPPE K, DEKKER M, et al. 2002. Capturing Chromosome ConformationJ. Science, 295: 1306-1311.
DOSTIE J, RICHMOND T A, ARNAOUT R A, et al. 2006. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elementsJ. Genome Research, 16: 1299-1309.
HAKIM O, MISTELI T 2012. SnapShot: Chromosome confirmation captureJ. Cell, 148: 1068.e1061-1062.
LIANG Z, ZHANG Q, JI C, et al. 2021. Reorganization of the 3D chromatin architecture of rice genomes during heat stressJ. BMC Biology, 19: 53.
LIEBERMAN-AIDEN E, VAN BERKUM N L, WILLIAMS L, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genomeJ. Science, 326: 289-293.
LIU R, XU R, YAN S, et al. 2024. Hi-C, a chromatin 3D structure technique advancing the functional genomics of immune cellsJ. Frontiers in Genetics, 15: 1377238.
MIAO J, FENG Q, LI Y, et al. 2021. Chromosome-scale assembly and analysis of biomass crop Miscanthus lutarioriparius genomeJ. Nature Communications, 12: 2458.
WANG D, XIAO S, SHU J, et al. 2024. Promoter capture Hi-C identifies promoter-related loops and fountain structures in ArabidopsisJ. Genome Biology, 25: 324.
ZHAO Z, TAVOOSIDANA G, SJöLINDER M, et al. 2006. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactionsJ. Nature Genetics, 38: 1341-1347.