聚类分析是一种无监督学习方法,旨在将数据集中的样本按相似性或距离划分为不同的组(即"簇")。与监督学习不同,聚类分析没有预定义的类别标签,而是通过算法在数据中发现潜在的结构。这种方法广泛应用于图像识别、客户分群、市场细分、生物信息学等领域,可以帮助我们揭示数据中的模式和关系。
本章将深入介绍聚类分析的核心概念、常用算法及其实现方法。我们将探讨几种经典的聚类算法,包括K均值聚类、层次聚类和均值聚类,了解如何通过 R 语言对数据进行有效的聚类。此外,还将演示如何对聚类结果进行评估和可视化,以帮助我们更好地解释分析结果,进而为数据驱动决策提供支持。
一、样品的距离
在聚类分析中,样品间距离用于测量样品之间的相似性或差异性,帮助我们理解数据的结构并进行分组。R语言提供了多种方法来计算不同的距离,包括欧氏距离(Euclidean Distance)、曼哈顿距离(Manhattan Distance)、布雷-柯蒂斯距离(Bray-Curtis Distance)等。
**(1)**欧氏距离:欧氏距离是最常见的距离度量方式,表示样品之间的直线距离,适用于连续变量数据。定义如下:
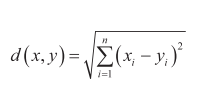
【例1】在R中使用dist()函数来计算欧氏距离。
在代码编辑器中依次输入以下代码,选中后单击RUN按钮运行。
data <- matrix(rnorm(25),nrow=5)
euclidean_dist <- dist(data,method="euclidean")
euclidean_dist运行后输出结果如下:
1 2 3 4
2 0.581547
3 4.153751 4.184982
4 1.364787 1.529577 5.099264
5 2.942890 2.988582 2.854306 3.302640**(2)**曼哈顿距离:曼哈顿距离也称为城市街区距离,表示沿坐标轴的路径距离,适用于一些离散数据或需要强调每个变量独立贡献的情况。定义如下:
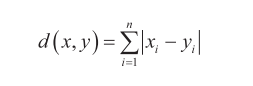
【例2】通过dist()函数指定method="manhattan"计算曼哈顿距离。
在代码编辑器中依次输入以下代码,选中后单击RUN按钮运行。
manhattan_dist <- dist(data,method="manhattan")
manhattan_dist运行后输出结果如下:
1 2 3 4
2 0.9706551
3 7.5949329 7.7510528
4 2.6860888 3.3497322 8.3663644
5 5.5287849 6.1406327 4.4447700 6.2353583**(3)**布雷-柯蒂斯距离:布雷-柯蒂斯距离主要用于生态学中,适用于相对丰度数据,用于测量样品之间的差异性。定义如下:
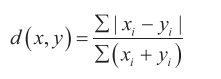
【例3】使用vegan包中的vegdist()函数计算布雷-柯蒂斯距离。
在代码编辑器中依次输入以下代码,选中后单击RUN按钮运行。
library(vegan)
bray_dist <- vegdist(mtcars[1:4,],method="bray")
bray_dist <- as.matrix(bray_dist)
bray_dist运行后输出结果如下:
Mazda RX4 Mazda RX4 Wag Datsun 710 Hornet 4 Drive
Mazda RX4 0.000000000 0.001237145 0.1347356 0.1440774
Mazda RX4 Wag 0.001237145 0.000000000 0.1340318 0.1428439
Datsun 710 0.134735626 0.134031813 0.0000000 0.2550549
Hornet 4 Drive 0.144077392 0.142843914 0.2550549 0.0000000**(4)**马氏距离(Mahalanobis Distance):马氏距离考虑了变量之间的相关性,适用于高维数据和有相关性的特征。它的计算公式为:
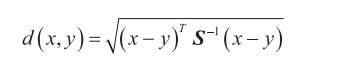
其中,S为协方差矩阵。
【例4】使用mahalanobis()函数计算马氏距离。
在代码编辑器中依次输入以下代码,选中后单击RUN按钮运行。
S <- cov(mtcars[1:6,1:4])
mahalanobis_dist <- mahalanobis(mtcars[1:6,1:4],
colMeans(mtcars[1:6,1:4]),S)
mahalanobis_dist <- as.matrix(mahalanobis_dist )
mahalanobis_dist 运行后输出结果如下:
[,1]
Mazda RX4 1.666667
Mazda RX4 Wag 1.666667
Datsun 710 4.166667
Hornet 4 Drive 4.166667
Hornet Sportabout 4.166667
Valiant 4.166667**(5)**闵可夫斯基距离(Minkowski Distance):闵可夫斯基距离是欧氏距离和曼哈顿距离的推广,参数p控制其形态。定义如下:
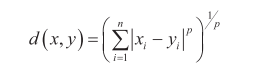
当p=1时,闵可夫斯基距离退化为曼哈顿距离,即每个维度差值的绝对值之和。
当p=2时,闵可夫斯基距离退化为欧几里得距离(Euclidean Distance),即常见的两点间的直线距离。
当p→∞时,闵可夫斯基距离退化为切比雪夫距离(Chebyshev Distance),即各维度差值的最大值。
【例5】在dist()函数中设置method="minkowski"和p参数求解闵可夫斯基距离。
在代码编辑器中依次输入以下代码,选中后单击RUN按钮运行。
minkowski_dist <- dist(data,method="minkowski",p=3)
minkowski_dist运行后输出结果如下:
1 2 3 4
2 0.5376354
3 3.5408809 3.5856678
4 1.1306428 1.1924417 4.5102983
5 2.4303189 2.4188221 2.6470566 2.7942379**(6)**杰卡德距离(Jaccard Distance):杰卡德距离是一种度量不相交集合之间相似度的距离,用于衡量两个集合之间的差异。它通常用于比较两组特征或标签之间的相似性,尤其是在生物信息学、文本挖掘和聚类分析中。
杰卡德距离的计算基于杰卡德相似系数。给定两个集合A和B,杰卡德相似系数定义为:
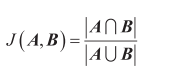
|A∩B|是集合A和B的交集元素个数,|A∩B|是集合A和B的并集元素个数。杰卡德相似系数的值范围在0和1之间,值越接近1,表示两个集合越相似,值越接近0,表示两个集合越不同。杰卡德距离是相似系数的补值,杰卡德距离也在0和1之间,值越接近0,表示集合越相似,值越接近1,表示两个集合差异越大。
杰卡德距离广泛应用于分类标签、二元特征或其他不相交集合的相似性比较,尤其应用在以下情况中:
文本相似性:计算两个文本的词汇集合的相似度。
生物信息学:比较两个生物样本中基因或特征的相似性。
聚类分析:用于基于集合的相似性进行聚类。
【例6】使用vegan包的vegdist()函数来计算杰卡德距离。
给定一个二进制矩阵数据,其中每行表示一个样本,每列表示一个特征是否存在,可以这样计算。
在代码编辑器中依次输入以下代码,选中后单击RUN按钮运行。
# install.packages("vegan")
library(vegan) # 安装包
data <- matrix(c(1,0,1,1,0,0,
0,1,1,0,1,1,
1,1,0,1,0,1),
nrow=3,byrow=TRUE) # 创建一个示例二进制数据集
jaccard_dist <- vegdist(data,method="jaccard") # 计算杰卡德距离
print(jaccard_dist)
运行后输出结果如下:运行后输出结果如下:
1 2
2 0.8333333
3 0.6000000 0.6666667在实际应用中,选择合适的距离度量方式对于准确的聚类分析非常重要。欧氏距离适用于连续变量数据,而布雷-柯蒂斯距离适用于生态学或相对丰度数据。通过结合不同的距离度量,可以更好地理解样品之间的相似性或差异性。
本篇文章讲了样品的距离知识点,接下来继续讲解层次聚类和均值聚类的知识点,关注我不迷路!
本文摘自《R语言医学数据分析与可视化》,具体内容请以书籍为准。