论文总结
1、作者提出了PIPE4,专为跨物种设计的快速蛋白质-蛋白质互作的预测引擎,解决大规模互作组预测的效率和精度难题。优化了相似性加权得分,归一化窗口频率,完美适配跨物种预测场景。
2、完成三类复杂预测:拟南芥→大豆跨物种、人 - HIV1 种间、大豆 - 大豆胞囊线虫(跨 + 种间)互作组。精度显著优于 PIPE3、SPRINT、SPPS 等主流方法;跨物种预测性能与进化距离正相关,近缘物种效果更优。
摘要
对更大规模且日益复杂的蛋白质-蛋白质相互作用(PPI)预测任务的需求,要求最先进的预测器高效且适应跨物种预测。此外,生成综合相互作用组的能力使得在所有预测背景下评估每个PPI成为可能,从而进一步提升了在极端类别不平衡情况下的分类性能,利用互惠视角(RP)框架实现。我们在这里描述PIPE4算法。PIPE3/MP-PIPE序列预处理步骤的适应使得速度提升了50倍以上,新的相似度加权评分在应用于任何跨物种和跨物种预测模式时,能够适当归一化窗口频率。生成三种预测模式的综合相互作用组:(1)跨物种预测,其中拟南芥作为预测综合Glycine max相互作用组的代理,(2)智人-HIV1之间的物种间预测,以及(3)结合跨物种和跨物种预测的模式,其中拟南芥和秀丽隐杆线虫作为代理物种预测大豆属甘氨酸max与甘氨酸异端线虫(大豆囊线虫)。将PIPE4与最先进的方案进行比较后,性能有所提升,表明它应成为复杂PPI预测模式的首选方法。
引言
阐明蛋白质间相互作用(PPI)网络是分子生物学研究的核心。对于构建细胞过程的机制模型,PPI网络还带来了诸如基因功能预测、疾病基因鉴定以及药物发现等挑战5,6。计算PPI预测技术已被开发出来,以补充和指导湿实验室实验工作。过去十年,PPI预测器的计算需求在规模和复杂度上均有所增加。随着高性能计算基础设施和算法优化的出现,预测综合相互作用组(即所有可能的成对PPI在蛋白质组内或蛋白质组之间的集合)直到最近才成为可能。虽然在实现方法上各不相同,PPI预测工具通常利用已知PPI集合(此前已通过经典湿实验室技术确认)中的信息,来判断是否有两个查询蛋白会物理相互作用。任何一种方法的实用性和可扩展性都取决于其所利用的信息。基于结构的方法在极端情况下需要对每个蛋白质进行三维(3D)表征,因此蛋白质组覆盖率较低。虽然有助于确定高度特定的PPI网络,但许多方法需要基于模板的建模,这往往计算量较大。此外,即使拥有生物体蛋白质组中每个蛋白质的完整三维结构信息,阐明单一假设PPI所需的计算时间复杂度也使得这些方法在中等规模的网络之外难以实现。在另一极端,基于序列的预测器仅依赖原始序列数据,因此适合研究全蛋白质组网络。此外,这些方法通常效率很高,单个PPI可以在一秒内预测。物种内的首次全面预测通过基于序列的方法报道了多种生物的相互作用组11,12。特别是蛋白质-蛋白质相互作用预测引擎(PIPE;最新版本MP-PIPE11,为清晰起见于本研究中标示为PIPE3)一直是这些进展的前沿,并促成了包括智人、酿酒酵母、秀丽隐杆线虫、拟南芥、果蝇、肌肉疣等多种生物的综合相互作用组的阐明。 以及甘氨酸最大3,11--17。
预测模式的规模和复杂度的增加
全面的物种内相互作用组对于阐明特定生物体内的分子生物过程非常有用。研究充分且拥有大量先前验证的PPI的生物体适合进行这些分析,而研究不足且缺乏可用来训练预测因子的生物则存在问题。此外,许多研究不足的物种对人类健康至关重要,阐明完整的相互作用组对于研究疾病发病机制(如寨卡病毒)至关重要18。我们在这里呈现了一个改进版的PIPE以应对这些挑战。除了深入了解生物体内的细胞过程外,PPI预测因子对生物体间网络(即跨物种)的应用还促成了疾病发病机制的确定19--21、新型药物的开发以及对相互作用组进化的见解22。本研究聚焦于预测极大相互作用组的计算挑战,以及定义结合跨物种和跨物种预测模式的最佳实践,以预测那些通常不适合生成综合相互作用组的生物体。
跨物种预测。研究不足且实验验证PPI极少的生物,如G. max,由于训练数据不足,不适合研究综合相互作用组。为了规避这一问题,可以让一个进化上相似且研究充分的生物作为代理。也就是说,使用经实验验证的代理物种内PPI来训练PPI预测变量;随后对研究不足的目标生物的蛋白质组进行预测。由于已知G. max PPI的可用性有限,我们这里使用拟南芥作为研究充分且进化相似的代理指标,以生成这些跨物种预测,如图1A所示。 跨物种预测。虽然PIPE在应用于物种间预测任务时已初步取得成功19,但该问题要求评分函数(可能解释短且连续亚序列的频率)是合适的,因为蛋白质组大小在不同生物体间差异较大,我们预计相似亚序列的数量也会大幅差异。该模式(见图1B)要求两个生物体之间PPI的预测分数,通过该窗口在蛋白质组中的流行率对该窗口的证据进行归一化。本研究描述了解释相似度加权评分的相关性。 预测相互作用组大小。历史上,PIPE被应用于越来越大、越来越复杂的PPI预测问题。最初,PIPE设计用于预测单个物种内少量的对鱼。它逐步优化以预测以蛋白质组大小为n、n(n + 1)/2的三角形数为扩展的综合相互作用组。在原始生物酿酒酵母中,蛋白质组大小为6,700≈,这包括一个包含2200万个独特预测PPI的综合相互作用组。经过分布式计算的大量优化,PIPE应用于智人,达到n≈21,000,构成了一个比人类多一个数量级的相互作用组:2.2亿。现在,蛋白质组G. max为n≈75,700,我们以更高一个数量级的水平运作,拥有28亿次物种内预测。为了快速及时计算此类复杂相互作用组,PIPE预处理数据表示方式如方法部分所述进行了调整。综合起来,适当组织这些日益复杂的预测模式,有望更好地预测这些极具价值的整体相互作用组。例如,尽管大豆豆科植物、G. max和大豆囊线虫(SCN)甘氨酸异端是研究不足的生物,我们希望识别出可能扰以保护作物产量的可操作质子泵抑制剂。幸运的是,A. thaliana和C. elegans分别是进化相似且研究充分的模式生物,能够作为这两种感兴趣生物的代理。这一预测模式涵盖了跨物种和跨物种的PPI预测,如图1C所示。最后,高通量预测器使得预测所有可能相互作用的全面集合成为可能,从而产生了上下文:能够评估给定的PPI预测相对于所有可能相互作用的能力。将PPI的倒惠视角(RP-PPI)级联机器学习层应用于这些数据,显著提升了在极端阶级不平衡情况下的预测性能16。作为适用于任何PPI预测器的元方法,我们进一步验证了RP-PPI在跨物种预测模式中的应用。
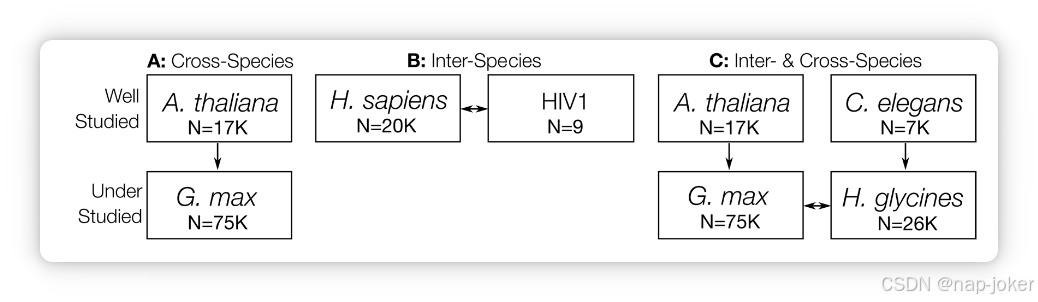
图1。跨物种预测模式。(A)跨物种模式,其中一个研究充分的生物的物种内PPIs可以作为代理指标,预测另一个研究不足但进化上接近的生物的综合相互作用组。(b)典型的物种间预测模式,利用两个研究充分的生物体内和/或两个生物之间的PPI来预测两者之间的综合相互作用组,以识别新的假定PPI。(C)结合跨物种和物种间预测模式,利用两种研究充分的生物的物种内PPIs来训练能够预测两个研究不足生物全面跨物种相互作用的模型。N表示蛋白质组的大致大小。
方法
过去十年,PPI预测任务的计算需求在规模和复杂度上均有所增加11,13,14,23。现有的PIPE实现MP-PIPE2于11年发布,为便于清晰起见,本文称为PIPE3,已足以完成这些任务。然而,全面预测G. max相互作用组需要对2,871,417,871个假定PPIs进行评分,这比综合人类交互组的规模增加了十倍以上。我们在此介绍了一个更高效的PIPE版本,恰如其分地命名为PIPE4,能够处理涉及大豆的预测任务,涵盖跨物种和跨物种的预测任务。简而言之,基础的PIPE算法会检查每个蛋白质的滑动窗口,以确定它们是否与已知物理相互作用的蛋白质对序列相似。预处理步骤会统计蛋白质中序列相似的窗口,以加快预测步骤。
用于描述算法变更的数学符号。我们在此澄清用于描述后续PIPE算法变更的符号。加粗符号表示一个集合,如向量、列表或元素集合。下标针对物种间应用进行调整,并识别特定生物蛋白质组中的某种蛋白质。为了简化符号,适当时省略下标。除非另有说明,使用一种生物体的例子则隐含于另一种生物。上标用于识别某一连续亚序列在给定蛋白质中的起始位置。希腊符号(如σ、Φ、φ、Γ、γ)用于表示函数,在适当情况下,其大写符号(如Φ、Γ)用于指代函数,而小写符号(如φ、γ)则方便地表示该函数应用后得出的集合大小。按照一般惯例,竖条 |·| 用于表示给定集合的大小,而上横线符号 ⋅ 表示集合的平均大小。最后,虽然⊕符号通常用作异或函数,但这里我们将其用作串接算符。为了直观地可视化正文中使用的符号,图2展示了两个任意蛋白质pai和pbj之间的比较示例,其中这两个窗口与每个生物蛋白质组中的所有其他窗口进行比较。在这种情况下,类似蛋白质的集合被证明是不相交的,正如跨物种和跨物种预测中所预期的那样;然而,对于物种内预测模式,我们可以预期相似蛋白质集合中存在重叠。在这个例子中,这对窗口(即交叉点的大小)的"命中"次数为6,这个值按照正文描述进行了规范化。 预处理蛋白质序列以实现窗口相似性。我们首先描述PIPE3的预处理步骤,即识别给定蛋白质组内相似的亚序列。我们认为生物a的蛋白质组大小为n,并且具有蛋白质名称列表(以整数索引表示),p = p , p , ..., p , ..., ..., p a a1 a2 ai an ,以及氨基酸序列列表 sa = sa1, sa2, ..., sai, ..., san,对应 p → s a a 表示pai具有序列sai。同样,具有蛋白质组大小为m的生物b具有=⣮ ⣰......Δ С� p p , p , , p , , p b b1 b2 bj bm , and s = s , s , ..., s , ..., s b b1 b2 bj bm 与 p → s b b b。我们记任一s的氨基酸序列长度为k(→即a、b的s与k为k)。长度为l的连续氨基酸序列,其中l≤k,属于s,记作"窗口"w。沿序列sai应用长度为l的滑动窗口,允许重叠并每次移动一个氨基酸,生成一组窗口列表:= ... ...− + w w , w , w , w , w ai 1 2 h (k l) 1 。为了澄清符号,生物a蛋白质组中蛋白质pai的任意窗口(序列sai和长度kai)以waxi表示,蛋白质pbj类似地称为wbj y。 使用相似函数 σ(w , w) 参考查询 ,用于判断蛋白质组中任意两个窗口是否被视为"相似",该窗口由通过 PAM120 氨基酸替换矩阵和相似阈值 τ 得到的值 v 定义。该函数接收两个窗口作为输入,第一个 wref 是来自一个蛋白质组的参考窗口,第二个 wquery 是另一个蛋白质组的查询窗口。如果窗口相似,该函数输出包含 wquery 的蛋白质索引。此外,该函数定义为支持窗口向量作为输入(用加粗字强调),并返回来自其他蛋白质组的对应蛋白质集合,其中相似:
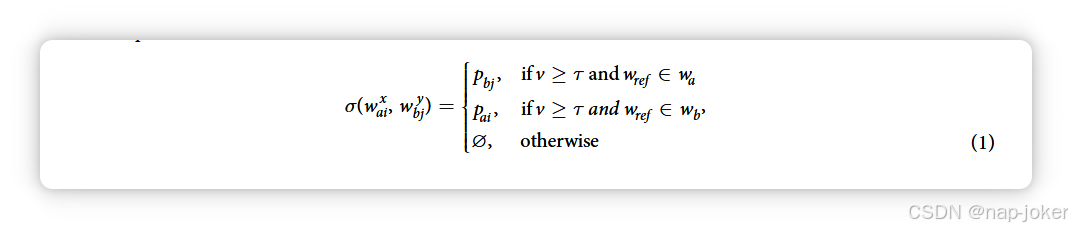
需要注意的是,来自蛋白质组a的窗口会返回蛋白质组b的蛋白质索引,反之亦然。每个窗口 waxi 和 wbj y 都有其对应的蛋白质列表 Φ,长度为 φ,且至少有一个窗口与之相似(φ ≥ 1)。对于长度分别为φaxi和φbj y,这些列表记为p , p , , p axi bj bj 1 b Φ = + ...φ 且 Φ = + ...φ p , p , , p bj y ai ai 1 a 。为了高效存储这些窗口相似性,每个蛋白质都写成一个数据库文件d,由无符号整数组成的杂乱数组,代表与窗口相似的蛋白质索引。任意蛋白质p的数据格式为:⊕φ ⊕Φ ⊕...⊕φ ⊕Φ −+ −+ dp: k 1 1 (k l) 1 (k l) 1 (2) 对每个生物体蛋白质组中的所有蛋白质进行此预处理步骤,生成一个数据结构,使预测完整相互作用组时能够实现恒定时间访问。对该预处理步骤中单对蛋白质(其中过杆表示平均值)的计算运行复杂度分析得到:O(k k φ ) (3) ab。这里,ka和kb分别是物种a和b的平均序列长度。预处理仅运行一次,且其运行时间比PIPE算法的其余部分少几个数量级,因此被视为固定的启动成本,因此在分析剩余的PIPE运行时可以忽略不计。

景观生成算法。对于任意一对蛋白质,pai 和 pbj,我们生成一个代表它们物理相互作用可能性的分数。得分足够高,超过某个全球定义的决策阈值,则获得积极分类。任意对的得分是对一个矩阵 ∈ ⁚ × M kai kbj 的矩阵应用聚合函数的结果。景观生成算法可以总结为:为了确定位置(x, y)的景观值,检查两扇窗 waxi, w bj y,并统计涉及具有相似窗口的蛋白质的已知相互作用数量。 算法1展示了该过程的实现,图2作为额外的图示。为清晰起见,我们定义了相互作用者函数 Γ(·),其输入为蛋白质索引 p,返回包含所有已知与 p 相互作用的蛋白质的长度列表γ。例如:
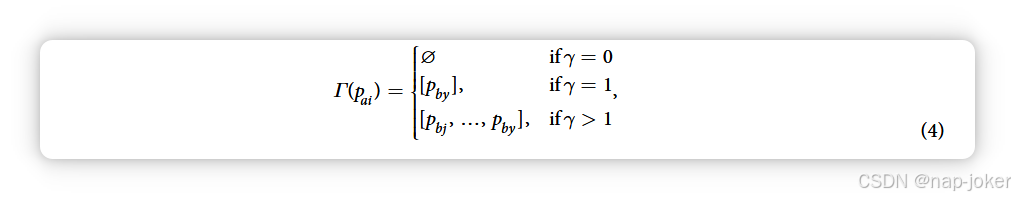
对PIPE地形生成算法(OL)计算复杂度的分析得出:
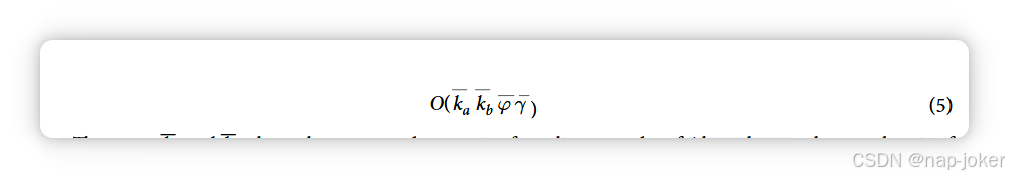
ka和kb这两个项通过对应关系,源自算法1的第2行和第3行,其中考虑了蛋白质pai和pbj中的每一对窗口。由于这些窗口重叠,窗口数量与每个蛋白质的长度成正比。第5行和第7行分别表示给定窗口φ相似蛋白的平均数量,以及给定蛋白质的平均相互作用次数γ。注意,去除下标是为了强调平均发生在两个蛋白质组上。第9行和第10行包含O(1)时间集成员操作和矩阵增量。计算出给定PPI的地形后,通过应用聚合函数获得最终得分。当用于物种间预测时,这一功能尤为重要,是通过窗口在各自生物蛋白质组中的流行率进行规范化。 相似度加权评分。相似度加权评分(SW-score)最初是Catalin Patulea于2011年开创性研究中提出的。该评分最初相较当时传统的PIPE评分有显著提升,但在用于物种间和跨物种预测时,这一指标重新引发了重要意义。相似度加权函数λ(x, y)输入了横向矩阵中一个单元格的坐标,索引为x和y,形式为:
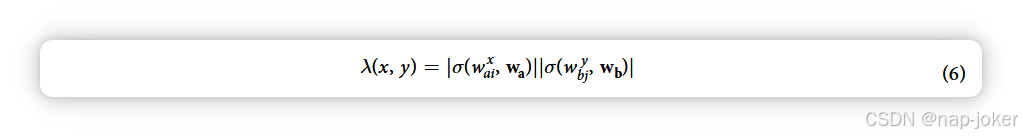
SW分数是对景观中所有单元施加λ(x, y)后,景观平均值的汇总值。该函数通过统计如果每对相似蛋白质相互作用时可能发生的相互作用数量,来规范某一点景观的高度。这抑制了与相互作用无关的高发窗口效应,并放大了相对罕见但频繁出现在已知相互作用中的窗口的影响。对于物种间应用而言,生物a、wa中所有窗口的集合可能与生物b、wb的集合有很大差异;而对于物种内应用,按定义W = w a b。 此外,在应用于跨物种预测时,训练数据可能会被多个生物合并,这种情况下由于各物种训练样本数量可能存在显著差异,这种规范化需要修改。在本研究中,我们考虑利用A. thaliana作为一个经过充分研究的代理指标,代表G. max进行跨物种预测,而目前可用的训练数据非常有限。同样,我们用秀丽隐杆线虫作为甘氨酸的代理,最终预测后两者之间的物种间相互作用组。将方程(6)简单应用于该PPI预测模式,使用下标A、G、C和H分别表示A. thaliana、G. max、C. elegans和甘氨酸的蛋白质组,得到
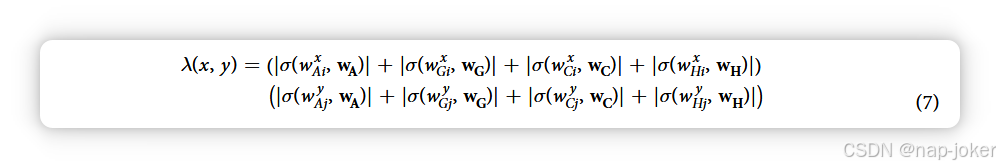
简化 A. thaliana 蛋白质组中 wAxi 窗口频率为 f = |σ(w , w )| Ai x Axi A,方程(7)变为:
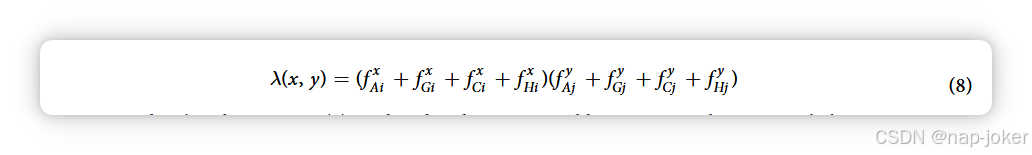
以此方式表述,式(8)意味着A. thaliana--G.max、A. thaliana -- C. elegans、A. thaliana -- 甘油杆线虫、C. elegans--甘油线虫和C. elegans--G. max之间可能存在相互作用,并将其归一化。虽然体外相互作用对这些生物的任何组合都可能,但训练数据中没有包含此类相互作用,这不公平地惩罚了与多个物种窗口相似的窗口。随着更多具有相似蛋白质的物种的加入,这种效应变得更加明显。此外,窗口不仅根据其在训练数据中的出现频率进行归一化,还取决于目标生物的蛋白质组中,而这些生物体的相互作用数据极少或完全没有。因此,归一化因子并不能反映训练数据中相似蛋白质之间可能相互作用的真实数量。我们在此提出了方程(8)的跨物种变体,仅在有可用PPI训练数据的物种中规范窗频率:
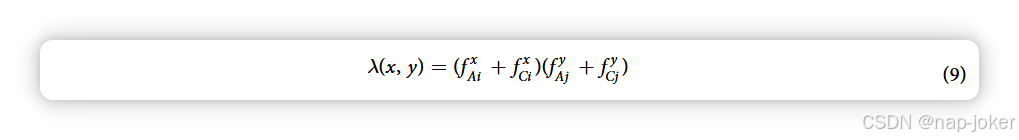
补充文件中描述了多项验证SW分数变化的实验。无论形式如何,将数据库文件加载到RAM中都可以实现对相应蛋白质组中类似窗口的恒定时间查找。计算假设PPI介于pa与pb(例如介于甘氨酸G. max与甘氨酸之间)的SW分数的复杂度,对每个细胞应用λ(x, y),随后的聚合得出平均运行时间为:
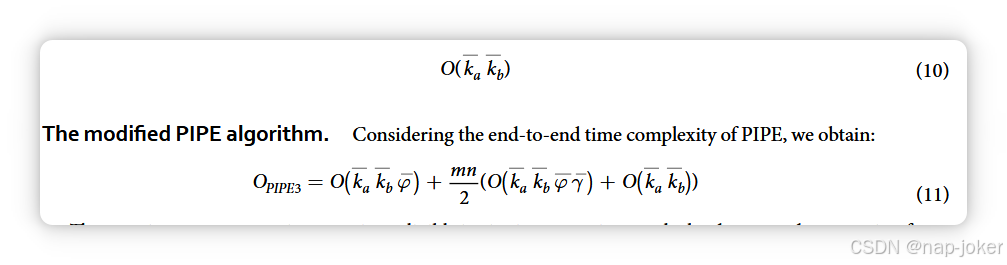
一次性预计算项的大小相比必须计算所有mn/2相互作用的景观函数和聚合函数项,可以忽略不计。由于目前需要将所有成对窗口都比较,ka和kb这两个项的限制,唯一可用的优化项是φ(给定窗口下相似蛋白质的平均数量)和γ(给定蛋白质的平均相互作用次数)。算法1的分析表明,第5至10行计算了单点的地形高度,可以重新表述为:给定窗口waxi和wbj y,计数轴Γ(σ(w, w))轴a和σ(w, w) bj y b的交点中蛋白质的数量:

其中,根据定义为φ ≤ m + n,通常为 φ m + n。因此,当每个蛋白质以 Γσw w(( , ))轴 a 环绕时,只有极少数会在交点中出现。对于任意一对集合,这些代价高昂的集合交集是不可避免的。此外,它们必须重复1/2(mn k k)a b次。为避免这种昂贵的交叉计算,提出了一种替代的预处理数据表示方式。为了直接预计算这些集合交集(以确保该交集的成员验证永远不会错误),我们首先希望生成一个具有key:value对的哈希表数据表示,其中键包含集合交集pψ的每个潜在蛋白质,而其对应值则包含该蛋白质在每个输入蛋白中出现的滑动窗口的索引列表。我们定义索引检索函数 Ψ(·),它以蛋白质为输入,返回一组蛋白质中出现的所有类似窗口的索引(注意加粗字体):
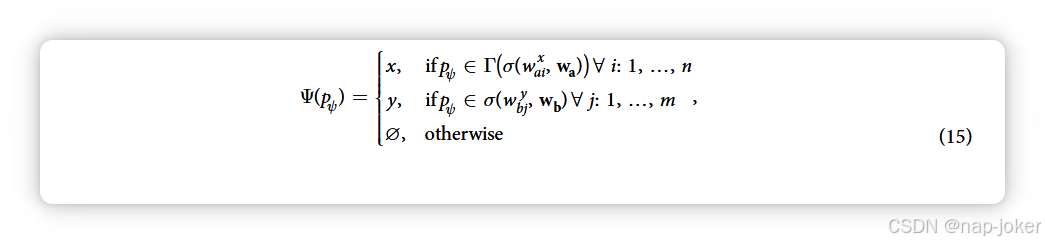
我们不存储蛋白质 pbj 中每个窗口位置 y 的蛋白质集合 σ(w , wb) bj y,而是存储 Ψ(pb),即所有包含 pψ 的集合 σ y 的集合。类似地,我们存储 Ψ(pa),即所有 x 的集合 Γ σ w w (((, )) 轴 a 包含 pψ 的集合。本质上,我们不是列出所有与某一窗口相似的蛋白质,而是列出所有与给定蛋白质相似的窗口,通过检查每个pψ并递增介于Ψ(pa)和Ψ(pb)之间的窗口对的景观,从而实现对每对窗口的共同子集(从而形成交互景观)。理想的结果是,我们从未执行过返回为假的成员校验,计算景观生成时间的预期减少为:
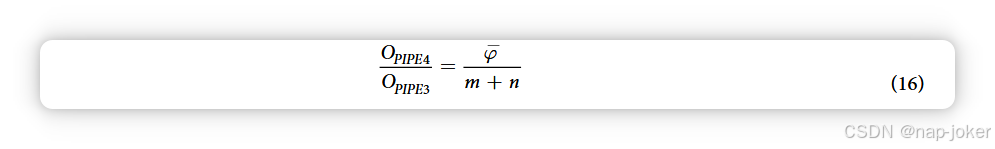
这里确定的PIPE4加速速度相较于之前的PIPE2算法有显著提升。实现这些修改需要两个新的数据库文件表示。为了简化符号,我们定义一个索引列表为 Ψ = Ψ(p ) ai ai,长度为 Ψai 。第一个数据库文件,记为d′w,格式与(2)中原始预处理版本相同,只是我们不是存储与数据库蛋白质每个窗口相似的蛋白质,而是存储蛋白质组中每个蛋白质的相似窗口。d′w 的格式如下:

结合原始景观生成复杂度OL,我们确定修正景观生成复杂度O′L为:
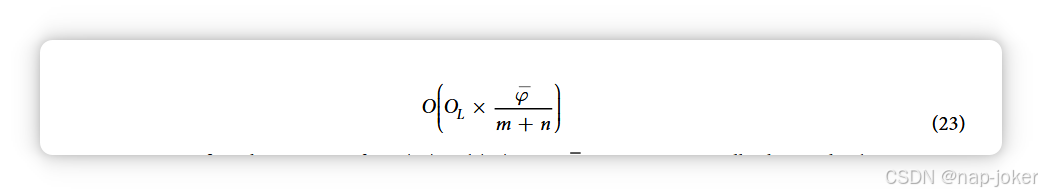
因此,我们确认(14)和(16)中的断言。由于φ m + n,新的景观生成算法预计在与参与 PPI 预测模式中生物蛋白质组大小成正比的速度下,速度将大幅加快。关于修改后的PIPE算法实验验证的更多细节,可参见补充文件。

预测大豆、人类-HIV1和大豆-SCN的综合相互作用组。为了预测综合相互作用组,我们需要已知的PPI数据和序列数据。所有PPI训练数据均来自BioGRID,交互数据被过滤为仅包含物理PPI。这些数据根据图1所示的三种图式整理而成:图1A获得了A. thaliana物种间的相互作用,图1B获得了H. sapiens与HIV1的物种间(仅)相互作用,图1C中获得了A. thaliana和C. elegans的种内相互作用。所有重复的互动都被删除了。智人、A. thaliana、秀丽隐杆线杆线虫和HIV1蛋白质组的氨基酸序列均来自UniProt数据库,仅选择了手动注释和审查的Swiss-Prot蛋白。如果BioGRID已知的相互作用与Swiss-Prot蛋白不对应,则该序列取自更大的TrEMBL UniProt数据库,该数据库包含自动注释和审查的蛋白质。G. max和甘氨酸螺旋桨的序列分别来自SoyBase和SCNbase。图1中定义的三个相互作用组随后通过上述PIPE4算法进行了预测。 多生物间及跨物种实验的数据集准备。本研究中涉及的每种生物体都需要一个高质量的已知相互作用PPI数据集。 对于每个生物体,我们从BioGRID25下载了构成物理PPI的物种内PPI数据(去除遗传相互作用、共定位或功能关联)。选择了物种内的PPI,以模拟一种生物的物种内PPI的使用,以预测另一种生物的物种内PPI。重复相互作用被从数据中剔除,最终过滤排除PPI含量为10个或更少的物种。符合该标准的十七种物种及其数据集组成列于补充表S4中。这些PPI数据集中每个蛋白质的氨基酸序列均从UniProt数据库下载26。使用了Swiss-Prot的人工注释和审查蛋白数据库,只有当BioGRID已知相互作用与Swiss-Prot蛋白不符时,我们才从更大的TrEMBL UniProt数据库(包含自动注释和评审蛋白)中提取该序列。 跨物种验证实验。PIPE完成跨物种PPI预测能力的初步演示在27年中展示了,利用酿酒酵母已知的PPI预测智人PPIs,反之亦然。虽然显示预测比随机概率更优,但跨物种预测的一般最佳实践未被进一步探讨。本研究的核心假说认为表现等级与进化距离等级呈反比;也就是说,由于蛋白质序列、功能和结构的进化保守,期望更近缘的物种在跨物种预测任务中表现更好。
一对多跨物种预测利用一个来源生物的训练模型,对其他目标生物进行预测,利用精确回忆曲线面积(AUPRC)和25%回忆(Pr@25Re)精度指标,考察进化距离与分类表现的关系。多对一跨物种预测利用训练好的多个生物池模型,对单个目标生物进行预测。在这些实验中,使用了肯德尔的Tau-b和斯皮尔曼秩相关检验,以考察相对进化距离是否与预测表现相关。每种生物的训练数据来自BioGRID,蛋白质序列来自UniProt;其具体组成见补充表S4。 进化距离与分类性能的关系。本研究的核心假说是,进化距离会影响预测表现,使得更近缘的生物能够更好地实现跨物种PPI预测。通过利用系统发育树估计每个生物体之间的进化距离,并将该距离与各条件的预测表现相关联来验证(见图3),但仅考虑了8个适合对2000个训练PPI进行随机子抽样(以控制训练数据量)的物种。特别值得注意的是,某些物种可能与其他物种进化距离相等,例如智人与M. musculus和R. norvegicus的进化距离均等。在这种情况下,我们期望得到的秩数是相同的。例如,在预测肌肉多粒虫PPI时,最高表现通常会在使用M. musculus PPI训练时,其次是R. norvegicus PPI,再是智人脉冲虫,依此类推。预测了这八种生物所有配对组合的综合相互作用组。使用10万对非交互对的随机子样本作为负集合,生成所有组合的ROC曲线并重复20次(以控制随机抽样程序);报告了平均ROC曲线。通过Spearman和Kendall的Tau-b秩相关检验测量进化距离与性能之间的相关性。Kendall的Tau-b秩相关检验和Spearman秩相关检验都被用来检验进化距离与性能(包括AUPRC和Pr@25Re)之间的相关性。在计算AUPRC指标的流行率校正精度时,我们选择了10:1(负:正)的类别不平衡。流行校正精度(PCPR)定义为:
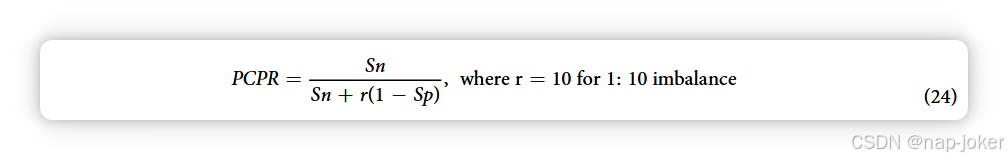
这方便地强制执行所选的类别不平衡,无论检测样本的正负数。类别不平衡会影响AUPRC指标的相对排序,但对Pr@25Re指标无影响。Kendall的Tau-b秩相关方程形式为:
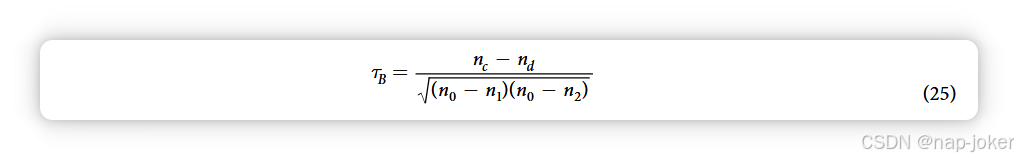
其中 nc 和 nd 是秩对一致和不一致的数量。这些分别计算预期和实际排位在一行中均匀大于或小的次数(一致)和不一致的次数(不一致)。除子对非重复秩对的数量进行校正,其中n1和n2是每列重复秩数,n = n(n − 1)/2 0 。在这里,我们通过独立计数每个测试物种的 nc 和 nd,然后对涵盖训练物种和测试物种对的单一 Tau-b 系数结果求和,调整了该相关系数。由于这种适应可能推翻传统Tau-b检验p值估计(特别是多重检验)的假设,我们与下文所述的基于置换的估计方法进行验证。统计显著性评估采用了两种独立方法。第一种(用R语言实现)近似了Tau-b和Spearman分布。第二种方法通过置换检验生成分布,进化排名在多次迭代中随机洗牌,计算每次计算的相关系数,所得p值为排名洗牌后产生相关系数大于或更极端的百分比。补充图中展示了智人(H. sapiens)的PR曲线示例。S5 并提出,那些与测试物种关系更为密切的生物,能提供更高的跨物种预测准确率。AUPRC和Pr@25Re实验的统计检验总结分别列于补充表S7和S8中。 跨物种验证实验。智人-HIV1跨物种结构涉及两个研究充分的生物,这些生物拥有足够的跨物种训练样本,以预测综合相互作用组。虽然大多数PPIs源自体外实验,但全面的物种间相互作用组对于揭示疾病发病机制和发现潜在药物靶点具有重要价值。鉴于人类与HIV1之间的较大进化距离,这一跨物种方案非常适合评估PIPE4相较于其前版本的性能。此外,这套预测对人体治疗药物的设计、开发和测试具有极大价值。将智人和HIV1的物种内PPI合并组成训练数据集。随后,利用先前发表的物种间PPI评估PPI预测变量的表现。阴性数据集包含一组随机抽样的PPI,其大小与已知阳性组大小相当。新PIPE4评分函数的性能与SPRINT PPI预测器、SPPS预测器、前PIPE3 PIPE-Score及原始相似度加权评分使用流行修正精度-回忆(PR)曲线进行比较。鉴于对人类-HIV相互作用组中阴性PPI的普遍性尚无共识,我们根据其他物种的先前估计并与其他实验保持一致,采用10:1比例。参见"与"最先进"的技术。人类与HIV1之间的综合预测被存放在Dataverse中,供更广泛的社区使用28。 互惠视角。如16中所述,RP-PPI元方法是一种级联机器学习层,利用基于上下文的特性提升PPI预测变量的分类性能。我们将它应用于本研究中生成的每一个相互作用组。为了验证RP-PPI方法用于跨物种预测,使用在一个物种上训练和评估的级联分类器,仅使用秩次和折叠类型特征对另一个物种进行预测,如16文所述;排除评分类型特征以控制生物体间的评分偏差。该测试针对五种生物的所有物种内数据集组合进行:智人、M. musculus、秀丽隐杆线杆线虫、酿酒酵母、A. thaliana。经过1000次自助迭代后,AUPRC相较基线PIPE4跨物种表现的平均增长被总结为热力图(参见"跨物种和跨物种预测的互惠视角"部分)。 比较PIPE4与最先进的PPI预测器。最后,我们将改进后的PIPE4方法与类似的PPI预测方法在计算速度、预测性能和所需计算资源(如所需内存)方面进行比较。虽然存在广泛的方法组合,但我们仅比较那些能够预测生物体内或生物体间所有可能相互作用对的方法,因此这些方法可用来利用RP-PPI方法提升预测性能。在之前的基准比较之后,我们考虑了Guo等人的方法29、Martin等人30、基于Shen等人的最新SVM方法"基于序列的蛋白质伙伴搜索"(SPPS)32,以及最近发布的SPRINT方法33。郭预测器使用支持向量机(SVM),利用一个特征向量,该序列包含七个物理化学性质的自相关值,这些属性为给定的蛋白质对。Martin预测器依赖特征向量,将序列信息编码在被定义为剔除子序列集合的签名乘积中;这些数据随后用于SVM中对PPI进行分类。SPPS方法是一种较新的基于测序的PPIs单向量模型预测器,其中蛋白质序列中编码的PPI信息被投影到联合三元组频率的向量空间中(作为一个单元研究氨基酸及其邻近氨基酸的物理化学性质);这些信息随后可用于SVM预测器。SPRINT方法使用间隔种子编码子序列中的相似性,然后处理这些元素以排除出现频率过高而无法参与交互的元素。根据SPRINT论文中基准测试结果,比较了Martin和Guo方法的性能,发现这两种方法的时间和内存需求(>2周计算时间和/或>256GB内存)过于过高,无法进一步考虑。这两种方法都无法在合理时间内预测整个人类相互作用组(~2.03亿对),因此也无法预测整个大豆相互作用组(~28亿对可能对)。此外,郭和马丁的方法此前曾与PIPE2方法进行比较,发现PIPE的祖先版本表现出更优的性能10。根据传递性质,比较PIPE4与Martin和Guo是多余的。基于这些原因,对于大规模综合相互作用组预测任务(例如大豆与SCN的比较),我们将比较范围限制在唯一已知能够预测综合相互作用组的另一种方法SPRINT。然而,对于规模较小的跨物种预测任务(人类-HIV1),我们将PIPE3、SPRINT和SPPS预测器与PIPE4进行了比较。这些实验在农业与农产品高性能集群BioCluster上进行,该集群由9台戴尔PowerEdge R930双插槽配置、Intel(R) Xeon(R) CPU E7-8870 v4(2.10 GHz)、1TB 1600 MHz内存、1.7TB SATA SSD组成。为了公平比较每种方法的运行时间,每个预测器被分配了20个并行线程,每个线程配备256GB内存。有关结果,请参见"与技术现状的比较"部分。
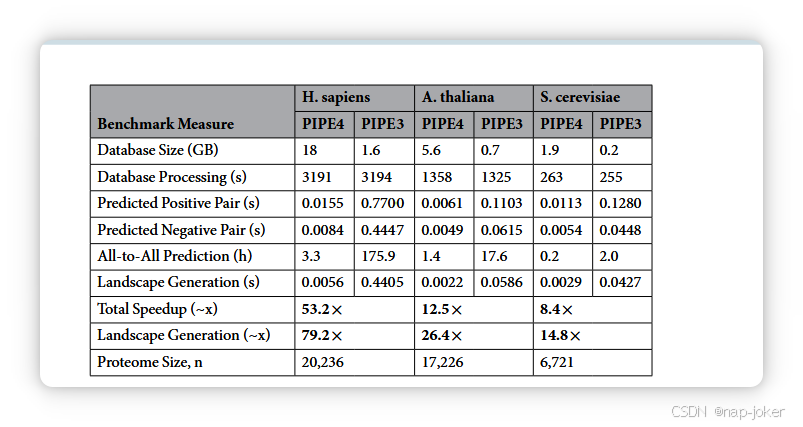
表1。中等规模簇的物种内基准测试结果。所有实验都使用18个节点,每个节点8个线程。
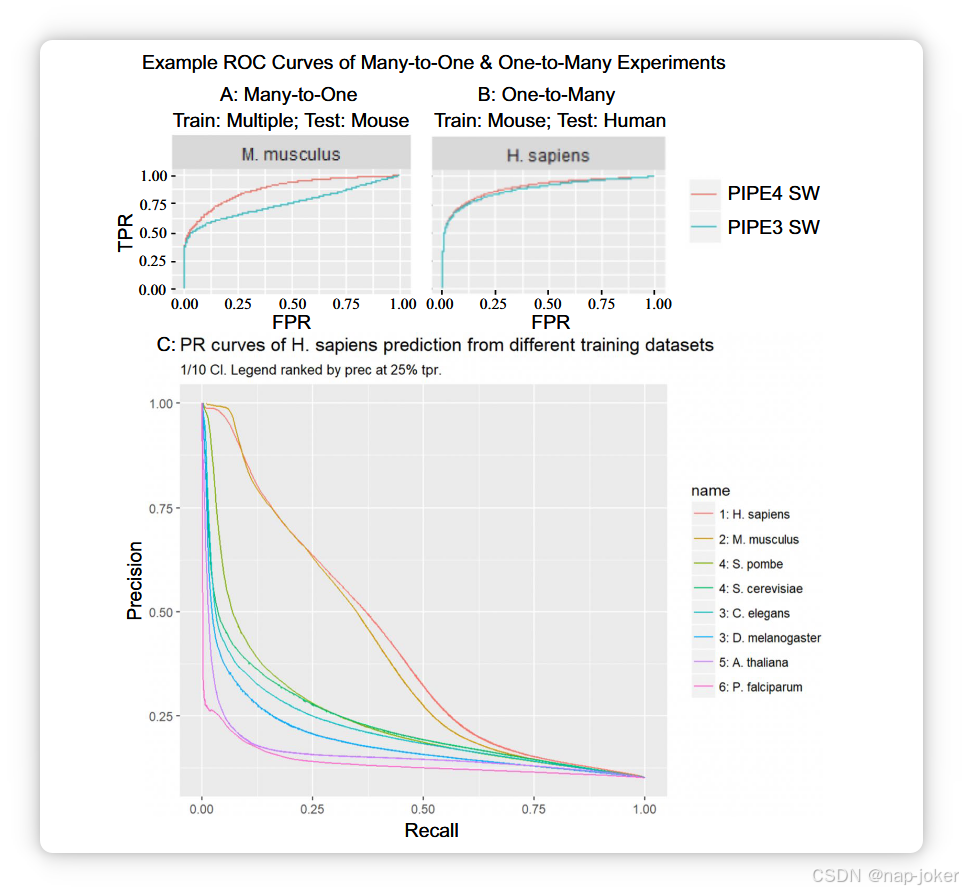
图4。多对一和一对多实验的ROC和PR曲线示例。(A) 描述了使用多种生物体评估小鼠PPI时的ROC表现。(B) 利用进化上近端的小鼠预测人类PPI表现ROC表现。(C) 在训练其他生物时预测人类PPI时的PR表现进行排名
结果
随着对越来越大规模和复杂的PPI预测模式的需求,现代算法必须适应以高效预测综合相互作用组。这里,PIPE算法的下一版本被调整用于跨物种预测,同时提升了计算时间复杂度和预测性能。据我们所知,PIPE4是首个专门针对复杂PPI预测模式开发的PPI预测器。除了其计算效率和PPI预测精度与最先进技术竞争外,PIPE还可以利用前述的PIPE-Sites算法提出假定的交互场地。Park的比较研究是对PPI在物种内、跨物种及跨物种模式中预测表现的首次评估之一。虽然当时仅考虑了人类和酵母,但研究中使用的PIPE2算法在考虑精度和回忆率方面表现优于竞争对手10。后续PIPE311旨在大规模扩展PIPE算法,用于综合预测任务(预测全蛋白质组交互网络),目前已进入第四次迭代,PIPE4算法已适配于物种间和跨物种的预测模式。在这里,我们希望在Park之前的发现基础上进行深入考察,深入探讨这些复杂模式中涉及的因素。 PIPE4加速实验。PPI预测令人尴尬地并行,因此总算法运行时间是计算单个PPI得分所需时间的函数。PIPE4算法用C语言编写,并利用MPI和OpenMP进行并行化,运行在一个中等规模的本地计算集群上。该节点由18个计算节点组成,每个节点包含100 GB SSD、32 GB内存和一颗Intel Core i7-3770 8核处理器,频率为3.40 GHz。将之前的PIPE3与当前的PIPE4方法在同一基准数据集上进行比较(通过物种内预测以适当评估每个版本),我们观察到结果与推导出的渐近复杂度一致(见表1)。也就是说,加速与蛋白质组大小之间的关系是线性的;预测的蛋白质组越大,加速越大。
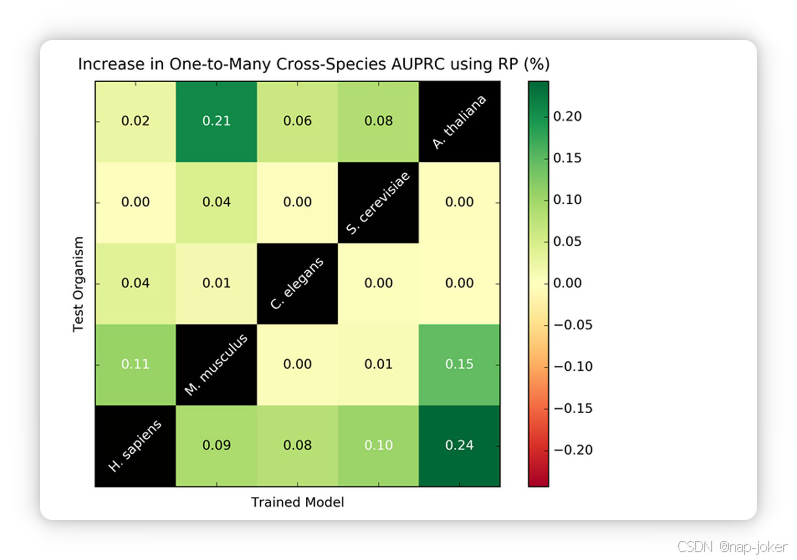
图5。利用PIPE4上一对多跨物种预测的AUPRC互惠视角增加。
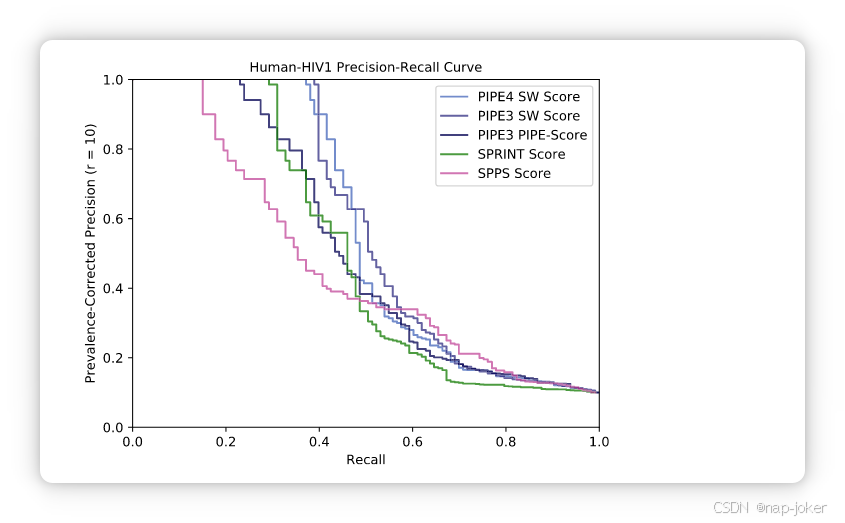
图6。比较PIPE3、PIPE4、SPRINT和SPPS之间的PR曲线与H. sapiens-HIV1物种间相互作用。
计算空间与时间的权衡导致数据库文件体积大幅增加。对应的数据结构必须能容纳在可用的内存中;然而,我们注意到,即使是最大的蛋白质组,这也不会超过所用机器的有限容量。由于绝大多数应用场景预计会在与人类蛋白质组大小相同或更小的蛋白质组上进行,数据库规模的增加是计算时间提升的合理权衡。此外,与现代基于序列的方法(如SPRINT)相比,PIPE4算法现在能够在类似基础设施下做出数量级的预测。 进化距离与分类的关系。对所有56种成对组合的一对多跨物种预测均采用AUPRC和PIPE3及PIPE4的 Pr@25Re 进行了总结。对均值差异的配对t检验显示,PIPE 4的平均准确率分别提升1.1%和1.9%的平均准确率(AUPRC和Pr@25Re,均值准确率均为1.1%和1.9%,均值均值差异为统计学显著(p < 0.001)(补充表S5)。因此,新的PIPE4评分方法在使用单一训练物种时显著优于PIPE3版本。同样,对8个群体组合的多对一跨物种预测进行了总结(补充表S6)。配对t检验显示出统计学显著差异(p < 0.01),AUPRC的平均准确率真实差异估计为9.6%,Pr@25Re为16.4%。由于修正后的SW分数支持多种训练物种的组合,我们考察了进化距离与分类表现的关系。在大多数情况下,进化距离与p < 0.05水平的性能显著相关(补充表S7,16/32;补充表S8,19/32)。从定性角度看,近缘生物在跨物种预测中表现良好,但之后表现会大幅下降(见图4)。这些发现表明,一对多跨物种预测适用于近亲和远亲物种,而多对一预测仅对非常近缘的生物有益。虽然未来在追求系统适用规则方面的研究是必要的,但有意进行自身跨物种和跨物种预测的研究者在准备训练数据集时,可以考虑本研究中的三个关键发现:(1)与测试物种关系更密切的生物,跨物种预测准确率更高。(2)一对多跨物种预测适用于近亲和远亲物种。(3)多对一跨物种预测仅对非常近缘的生物有益。 跨物种和跨物种预测的互惠视角。当RP应用于跨物种预测时,仅考虑等级型和折叠型特征;原始乐谱被排除在模型之外。因此,我们无法期待与物种内应用相比的性能提升。大多数调控/测试物种配对中AUPRC相较于PIPE4时呈非负增长(见图5)。性能提升最大的是PIPE4表现特别差的情况(例如训练A. thaliana和测试智人)且两者蛋白质组大小相近(如A. thaliana和M. musculus),表明它们可能共享相似的蛋白质相互作用分布。根据RP方法的定义,我们不期望性能提升与进化距离相关,而更多是蛋白质组大小和每个蛋白质相对PPI分数的秩序分布。蛋白质组大小最小的生物体不会显著提升蛋白质组数量较大的生物分类表现。因此,RP元方法最适合增强跨物种预测表现,通常可应用于跨物种模式,成功程度各异。尤其是对于初始分类器表现不佳的跨物种预测,RP可以显著提升性能;而初始分类器表现良好时,应用RP通常会带来适度的提升或相当的性能。 与技术现状的对比。比较PIPE3、PIPE4、SPRINT和SPPS方法在人类-HIV1物种间PPI预测任务中的表现,我们注意到SW评分在回忆值范围内提升了PIPE3 PIPE-Score和SPRINT评分的精度(见图6)。此外,PIPE4 SW分数在我们最期望的精度范围内占主导地位,即Pr≥ 0.5,其中至少一半的正面预测属实。PIPE3和SPRINT成绩在此范围内表现相似。SPPS方法在所有方法中表现最差。生成这些综合交互组所需的硬件需求在PIPE4和SPRINT之间相当。评估最大预测任务所需的RAM(G. max-H. glycines)分别使用~150GB:153和151。SPPS方法不适合本研究,因为它远远超出了RAM的限制;>250GB。PIPE4算法明确设计以支持跨物种和跨物种的预测模式。现有绝大多数基于序列的PPI预测器,包括PIPE3算法,都未考虑跨物种和跨物种预测。因此,在物种间的比较对竞争方法可能不公平。然而,与Park用PIPE210的发现一致,我们观察到PIPE3的SW分数始终优于其他竞争方法,且未考虑物种间的背景(见图6)。SPRINT和SPPS方法都可能从调整其算法以适应这些复杂预测模式中受益。 摘要。随着适用于复杂预测模式的PPI预测器越来越准确高效的应用,研究人员能够生成原本难以实现的综合交互组。例如,生成甘氨酸-G。MAX相互作用组能够为这些物种之间的分子机制(如宿主-病原体相互作用)提供前所未有的洞察,这可能对农业和经济产生深远影响。在这项工作中,我们引入了PIPE4算法,发现其速度显著快于前身PIPE3,智人、A. thaliana和酿酒酵母分别实现了53.2倍、12.5倍和8.4倍的加速。修正后的SW分数被证明能提升涉及跨物种和跨物种预测的复杂PPI预测模式中的表现。元方法RP被证明对这些复杂的跨物种预测模式有效。最后,如果竞争的PPI预测算法能够考虑训练样本的来源,预计它们在这些复杂模式中的预测性能将有所提升。我们发布了跨物种人类-HIV1预测的全对综合预测,以惠及科学界;可于 https://doi.org/10.5683/SP2/PVOTRN 获取。