据统计,超过70%的上市药物分子含有可离子化的官能团。pKa直接影响分子的溶解度、脂水分配系数、跨膜通透性(Permeability)以及与靶点蛋白的结合模式,是药物研发中的"灵魂参数"。
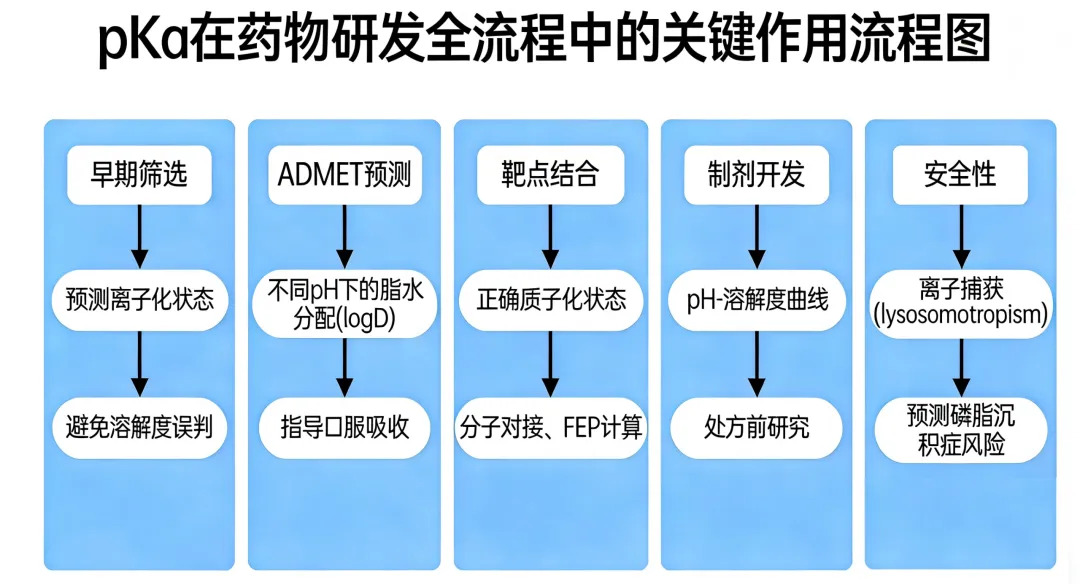
然而,精准预测pKa始终是计算化学领域的一大挑战,传统的经验公式(如 QSPR 模型)面对复杂、多氮杂环或高度官能团化的先导化合物时,误差往往超过 1-2 个数量级。量子化学方法(如DFT)则耗时数小时。如果 pKa 预测不准,将导致一系列连锁反应:溶解度误判、ADMET瓶颈、分子对接失效......
为了解决这一问题,InDraw 7.2在技术上深度集成顶刊《JACS》的最新科研成果------《Bridging Machine Learning and Thermodynamics for Accurate pKa Prediction》。实现了从"经验推断"到"热力学感知"的技术飞跃,下面我们用数据说话,看看它到底强在哪?

一、 精度验证:超越 Marvin等行业标杆,盲测平均绝对误差(MAE )仅 0.55
在科学界,数据是检验真理的唯一标准。新一代预测技术在多个国际权威数据集上进行了横向对比,结果令人振奋。
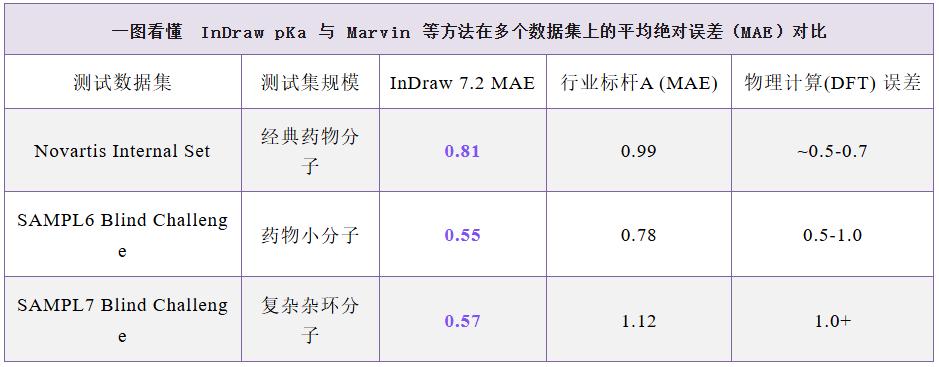
注:MAE < 0.7** 属于高精度模型,**MAE 0.7-1.0** 属于合格水平,**MAE > 1.0 则精度不足
药物分子集(Novartis Set)测试:在针对真实药物分子的测试中,预测模型的平均绝对误差(MAE)降低至 0.81 左右。作为对比,传统的行业标杆软件(如Epik或Marvin)的误差通常在 1.0 附近。这意味着预测结果更接近真实的实验反馈。
盲测挑战赛(SAMPL系列):在更具挑战性的 SAMPL6 和 SAMPL7 全球盲测数据集中,该技术展现了极强的泛化能力。在 SAMPL6 集中,MAE 仅为 0.55,在复杂杂环分子的盲测中,相较于传统的行业标杆软件,准确度提升了50%!这一数据已经触及了目前计算预测的精度天花板,表现甚至优于许多复杂的物理化学计算流程。
计算速度的降维打击:最令科研人员兴奋的是,在获得接近量子化学(DFT)计算精度的前提下,计算耗时缩短了数万倍。原本需要数小时的计算任务,现在只需 30 毫秒即可完成,真正实现了"即绘即显"。
二、为什么精准预测 pKa 如此困难?
在药物设计中,药化专家经常会遇到"预测值与实验值对不上"的困局,主要源于复杂分子内部微妙的化学环境:
诱导效应与共轭效应的叠加: 当分子中存在多个杂原子时,电子云的分布变得极度复杂,简单的加和法无法捕捉真实的解离倾向。
空间位阻与内氢键: 分子的三维构象会显著影响质子的解离难度。某些官能团可能被"屏蔽"在分子内部,导致其实际pKa远偏离理论值。
互变异构的干扰: 一个分子在溶液中可能存在多种异构体,每种异构体都有其独特的电离常数,如何计算其宏观观测值是业内的长期难题。
三、 技术解法:给AI装上"物理大脑"
InDraw采用Uni-pKa预测技术的跨越式升级,源于其独特的算法架构------将热力学循环约束与3D图神经网络(GNN)深度融合。
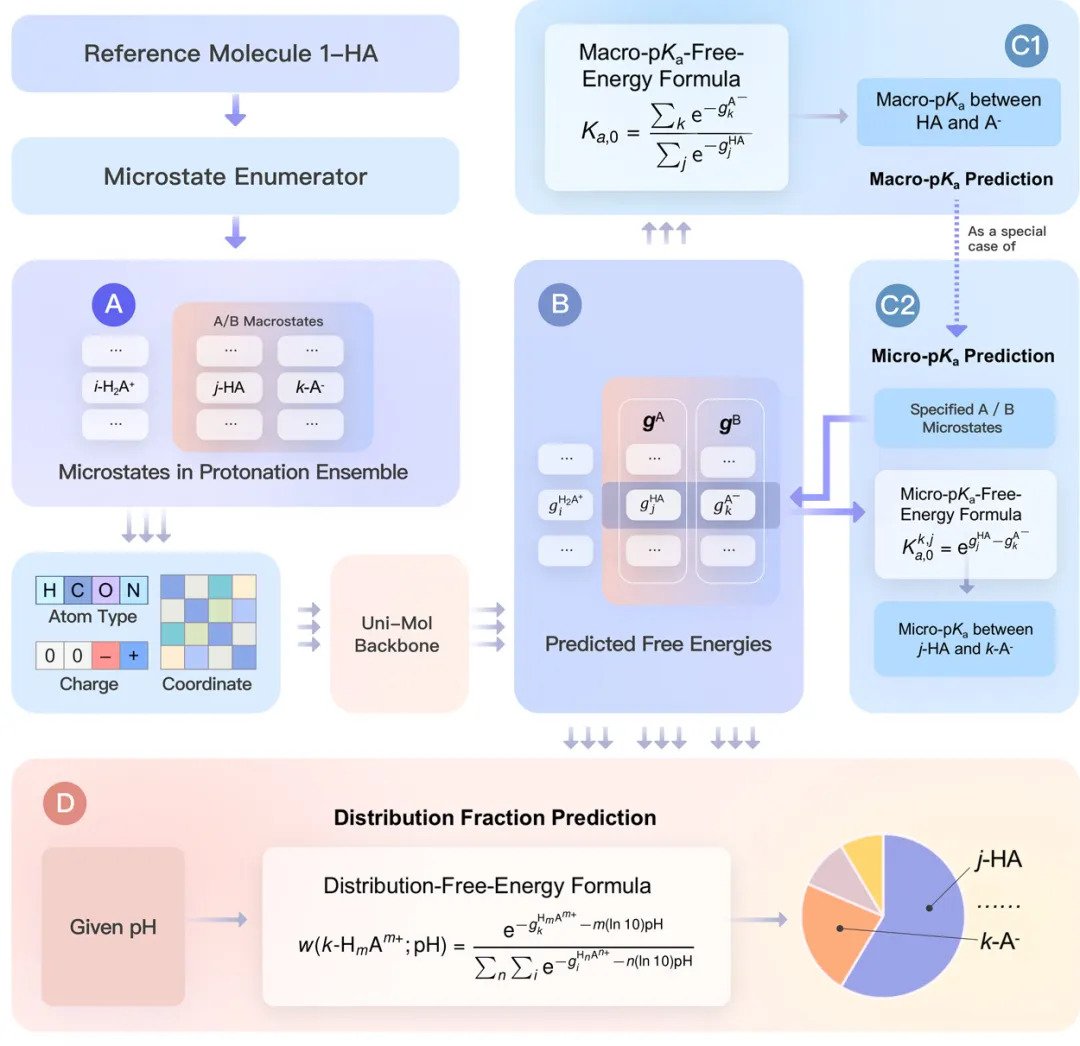
Uni-pKa 模型(如上图所示),通过 AI 学习分子的三维空间特征并预测各微观状态的自由能(Section B)。随后,利用严谨的热力学公式(Section C)将能量信息转化为精准的 pKa 数值。这种'物理规律约束 AI'的设计,不仅极大地提升了预测精度,更确保了复杂分子在多位点解离时的逻辑严密性。"
遵循物理法则的"系综"建模不同于市面上许多将pKa视为单纯"分类"或"回归"问题的AI模型,InDraw 7.2 采用了Uni-pKa的"质子化系综(Protonation Ensemble)"框架。该技术通过模拟分子在特定环境下的所有可能微观电离状态,并引入吉布斯自由能平衡约束,确保了预测结果在逻辑上严丝合缝。这意味着,无论分子结构多么复杂,其微观电离状态与宏观观测到的pKa值在物理学上始终是自洽的,告别了AI预测常见的"黑盒错误"。
空间感知的3D特征提取新技术不再局限于分子的2D连通性,而是通过预训练的3D分子模型,精准感知原子在空间中的相互距离与相互作用。无论是仲胺、叔胺的碱性漂移,还是多氮杂环的电荷分布,都能以毫秒级的速度给出极具信服力的解释。
四、体验升级:看得见的pKa,点得准的位点
InDraw 7.2 客户端将前沿技术转化为科研人员触手可及的交互功能:
交互式位点标注: 不仅给出数值,还能在分子结构上直观标注酸性/碱性位点,并根据强度进行颜色区分。这种直观的视觉反馈,能极大地辅助化学家快速进行取代基修饰。
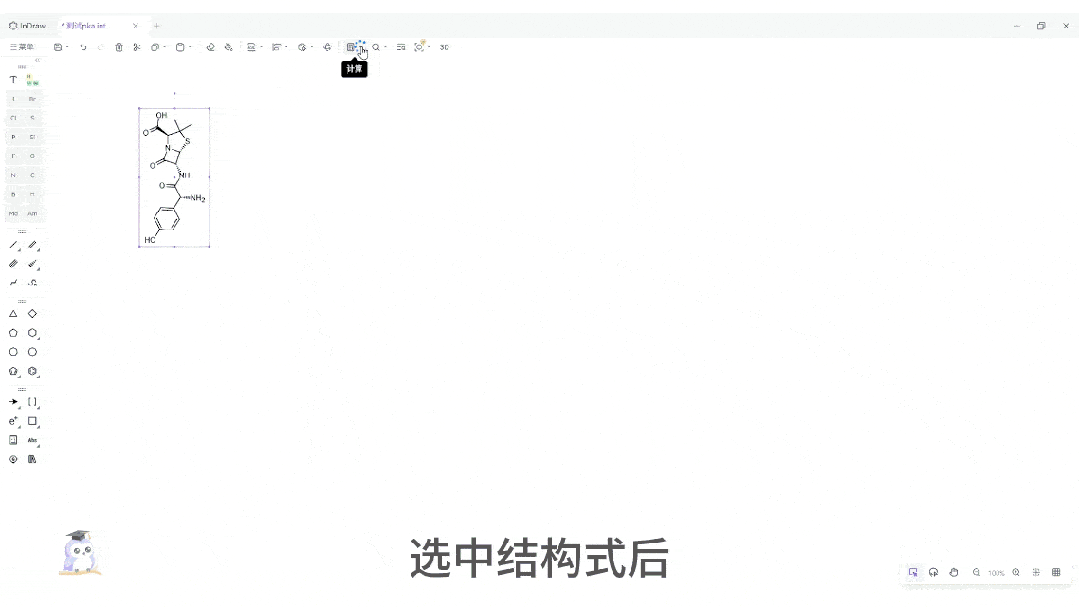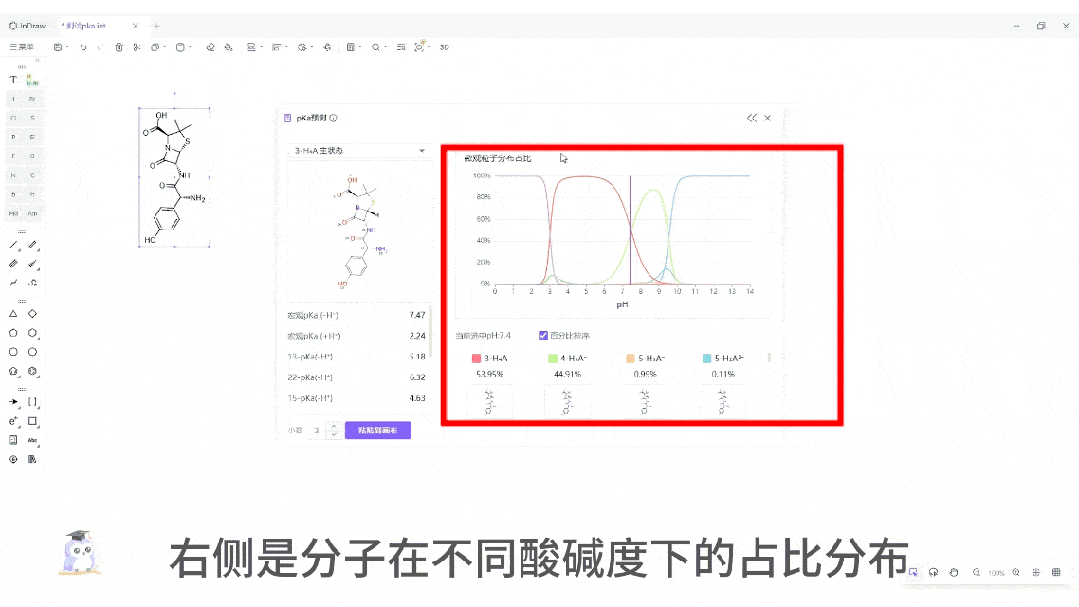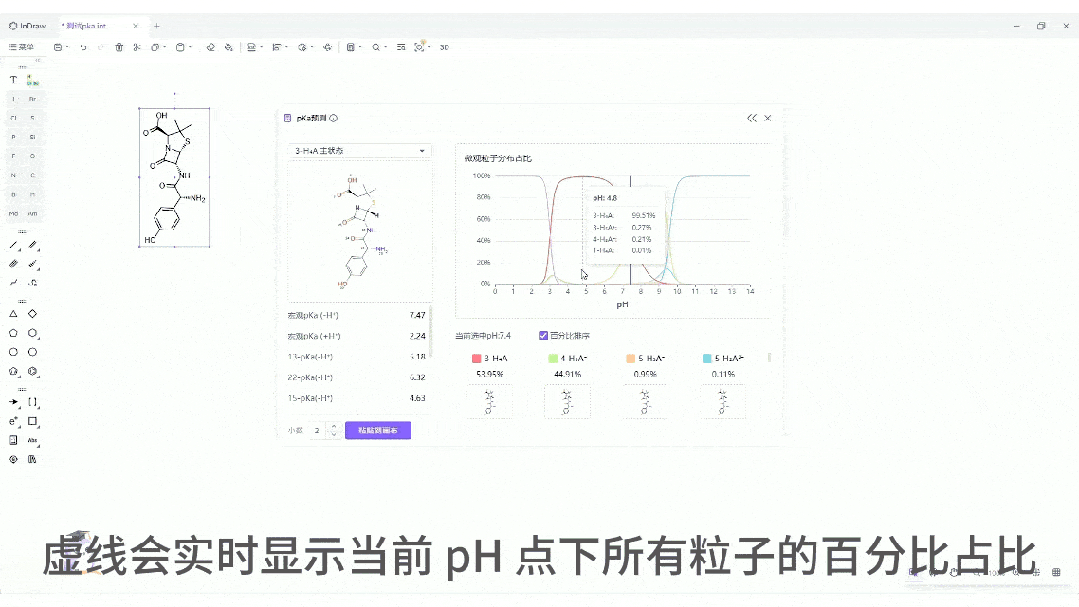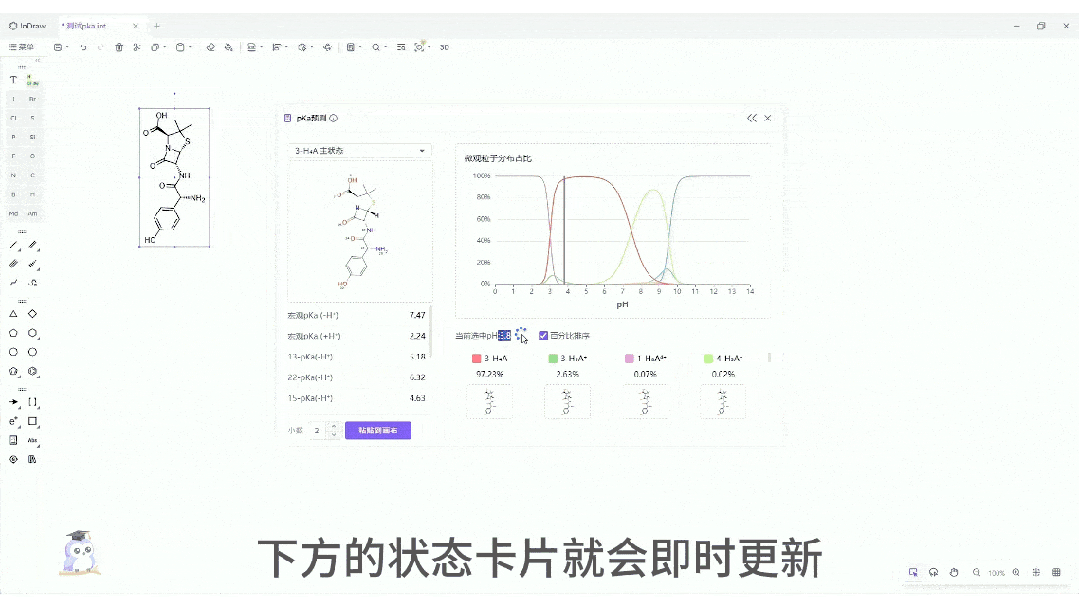
微观粒子分布曲线:一键生成分子在 pH 0-14 范围内的电荷状态分布图。这对于制药工艺设计、制剂处方前研究(Pre-formulation)具有至关重要的指导价值。
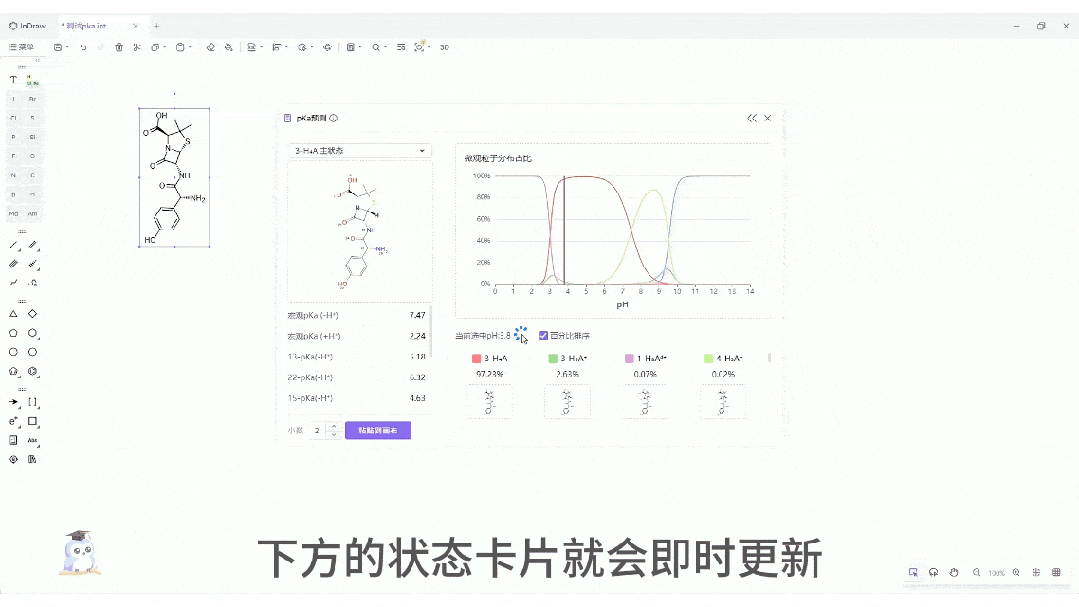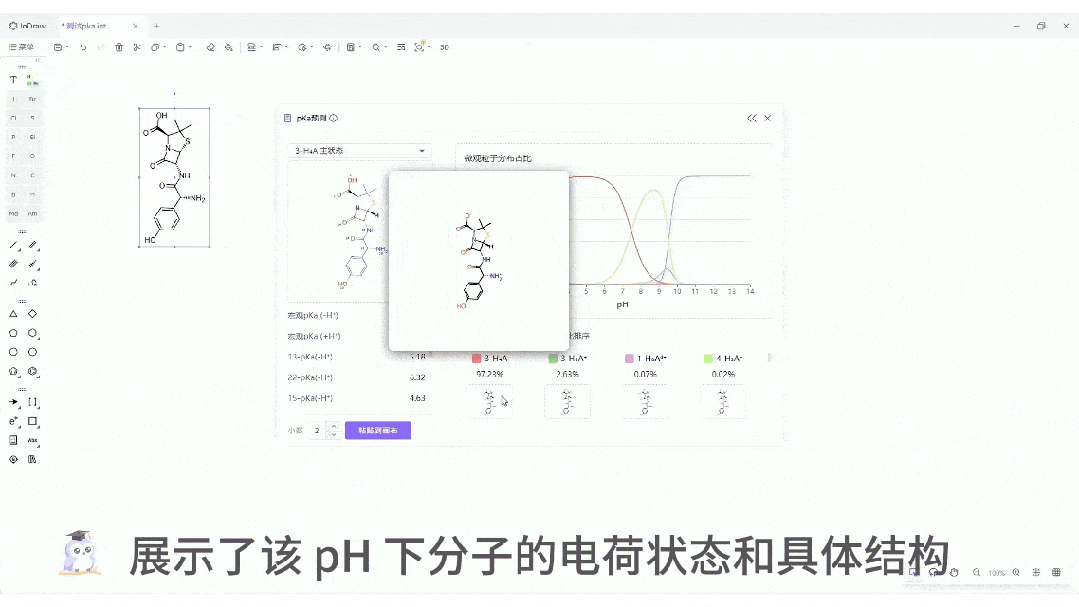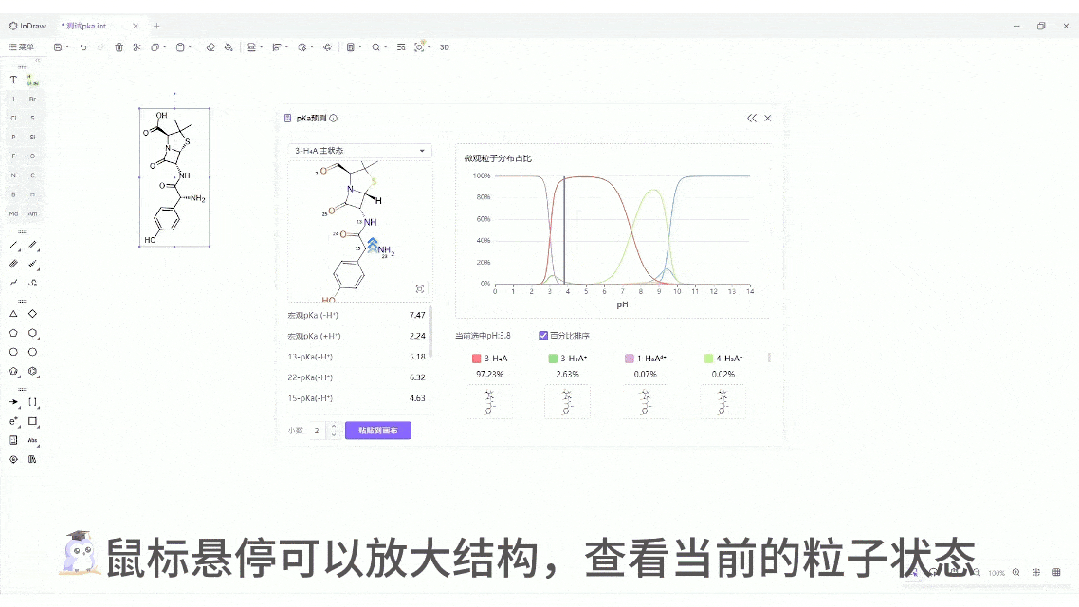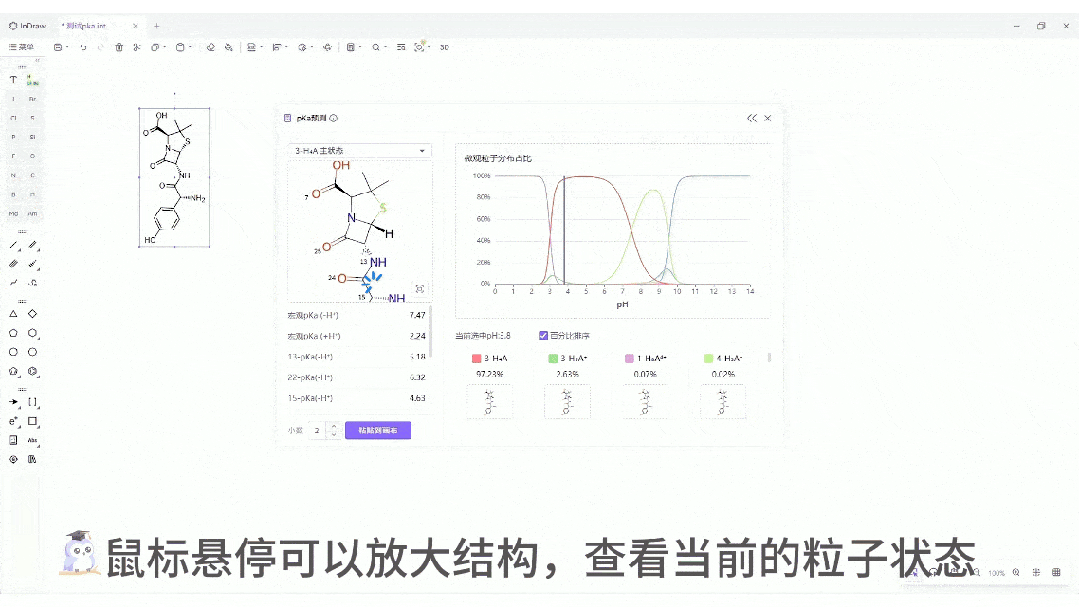
五、从"盲猜"到"秒答":改变化学家的工具箱
InDraw 7.2 的发布,不仅是绘图工具的更新,更是 InDraw 迈向"AI+化学智能工作站"的关键一步。科研人员不再需要反复猜测分子的理化性质,不再需要为了一个参数查阅无数文献,也不再需要等待漫长的量子化学计算排队。
下载并打开 InDraw 7.2,在指尖滑动的瞬间,强大的预测引擎便会为您揭示分子内部的电离奥秘。
参考文献:Luo, W., Zhou, G., et al. "BridgingMachine Learning and Thermodynamics for Accurate pKa Prediction." JACSAu 2024, 4, 3451--3465.