搞定生信数据的秘诀之一:是会使用工具!
工具用的好,就可以实现科技解放生产力,解放双手,节约时间!
实验室里最被导师看好的学生,一定是产出最高的那个人。
那如何高产出呢?百沐一下来帮忙!今天就来分享,如何秒出UMAP图。
一、降维聚类分析------生物科研人跨不过的坎儿
单细胞这么多的RNA数据,如何判断哪个是最有可能成为"病灶"的呢?
聚类分析的目的,简单来说,就是把复杂的问题归归类。
一张好的聚类分析图是开启下一步实验的基础。
在以前生成一张聚类分析图,不仅要搞定难懂的数据,还要从各种网站上下载R包。现在简单了,只需要百沐一下,你就知道!
二、给出指令
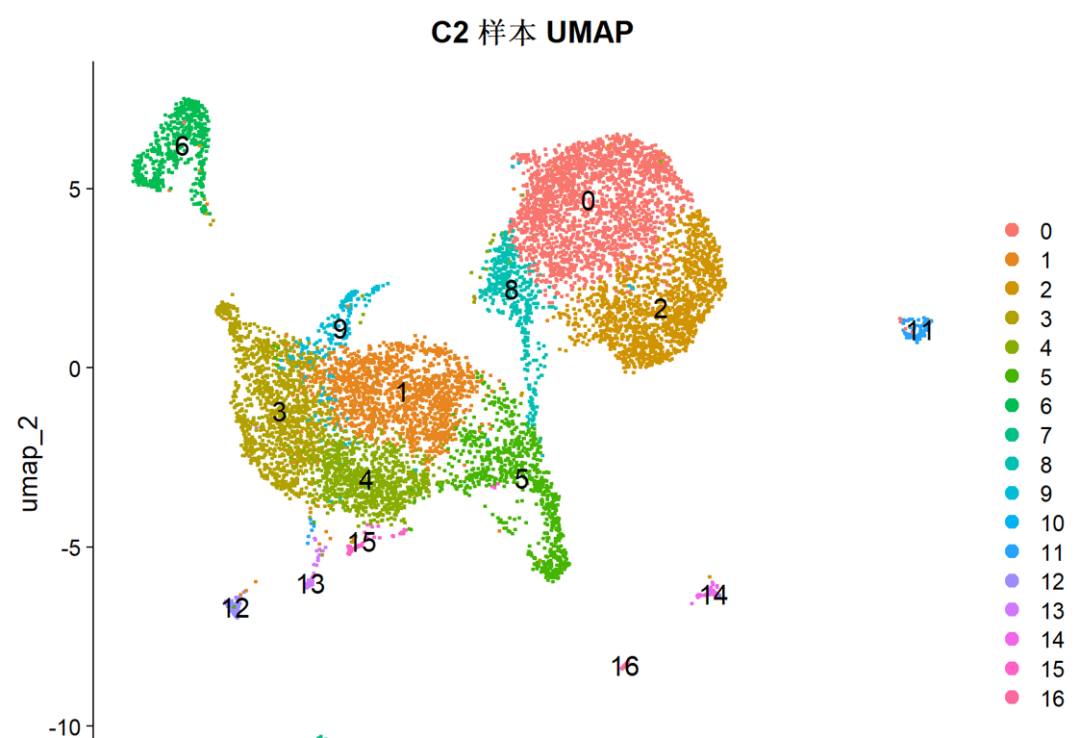
请你用R语言帮我分析C2这个样本,出一个UMAP图
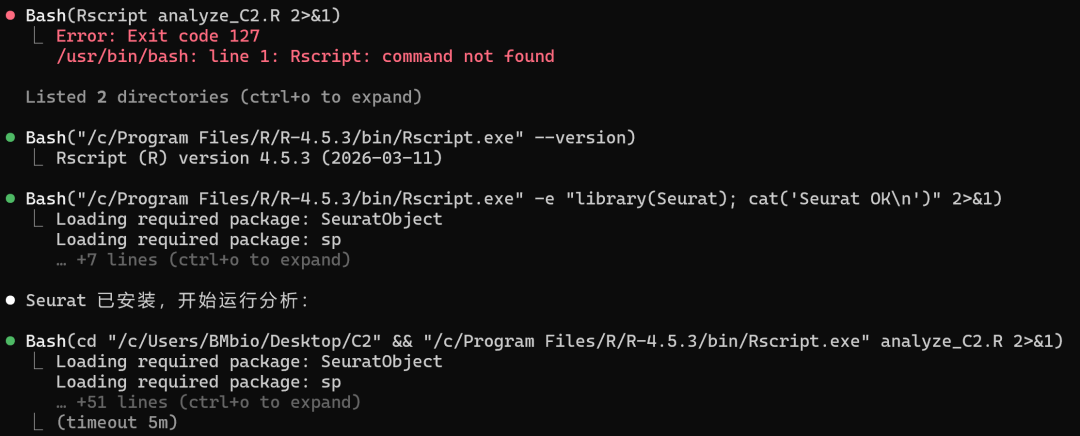
百沐可以自动为你写好脚本,并运行分析:
只需要五分钟就可以出图和结果:
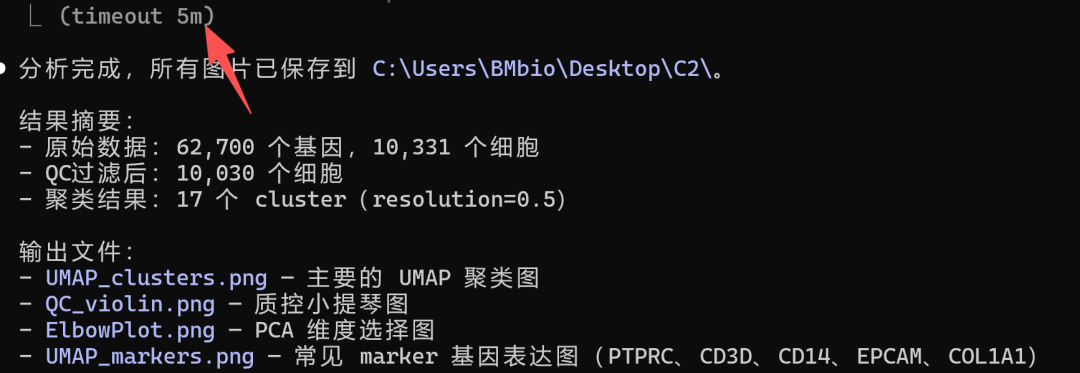
并且,这个样本的细胞类型也一并解决了!它会告诉你:
从 PCA 的 top genes 来看,这个样本包含成纤维细胞(COL1A1、LUM)、上皮细胞(KRT5、DSP)、内皮细胞(PECAM1、VWF)、免疫细胞(PTPRC)
等多种细胞类型,是一个组织样本。如果需要进一步做细胞类型注释,告诉我就行。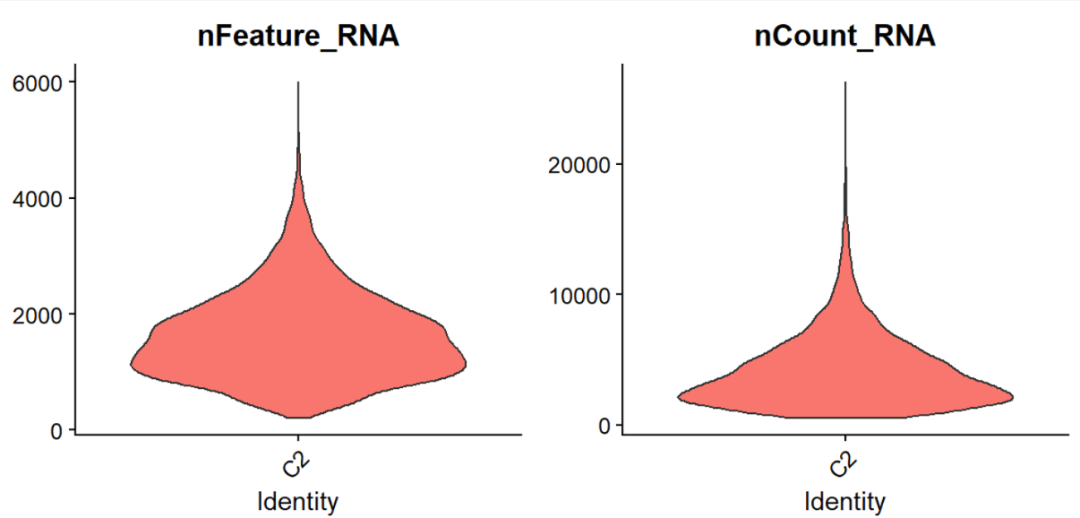
以下R包建议是收藏!
# C2 单细胞RNA-seq 分析 - UMAP可视化
# 使用 Seurat 分析 10x Genomics 数据
library(Seurat)
library(ggplot2)
library(dplyr)
# ---- 1. 读取数据 ----
cat("正在读取数据...\n")
data_dir <- "C:/Users/BMbio/Desktop/C2"
counts <- Read10X(data.dir = data_dir)
cat(sprintf("读取完成: %d 个基因, %d 个细胞\n", nrow(counts), ncol(counts)))
# ---- 2. 创建 Seurat 对象 ----
seurat_obj <- CreateSeuratObject(
counts = counts,
project = "C2",
min.cells = 3,
min.features = 200
)
cat(sprintf("过滤后: %d 个基因, %d 个细胞\n", nrow(seurat_obj), ncol(seurat_obj)))
# ---- 3. 质控 ----
seurat_obj[["percent.mt"]] <- PercentageFeatureSet(seurat_obj, pattern = "^MT-")
# QC 小提琴图
p_qc <- VlnPlot(seurat_obj,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3, pt.size = 0)
ggsave("C:/Users/BMbio/Desktop/C2/QC_violin.png", p_qc, width = 12, height = 4, dpi = 150)
cat("QC图已保存: QC_violin.png\n")
# 过滤低质量细胞
seurat_obj <- subset(seurat_obj,
subset = nFeature_RNA > 200 &
nFeature_RNA < 6000 &
percent.mt < 20)
cat(sprintf("QC过滤后: %d 个细胞\n", ncol(seurat_obj)))
# ---- 4. 标准化 & 高变基因 ----
seurat_obj <- NormalizeData(seurat_obj)
seurat_obj <- FindVariableFeatures(seurat_obj, nfeatures = 2000)
# ---- 5. 缩放 & PCA ----
seurat_obj <- ScaleData(seurat_obj)
seurat_obj <- RunPCA(seurat_obj, npcs = 30)
# Elbow plot 确定维度
p_elbow <- ElbowPlot(seurat_obj, ndims = 30)
ggsave("C:/Users/BMbio/Desktop/C2/ElbowPlot.png", p_elbow, width = 6, height = 4, dpi = 150)
cat("Elbow图已保存: ElbowPlot.png\n")
# ---- 6. 聚类 & UMAP ----
n_dims <- 20 # 根据 elbow plot 可调整
seurat_obj <- FindNeighbors(seurat_obj, dims = 1:n_dims)
seurat_obj <- FindClusters(seurat_obj, resolution = 0.5)
seurat_obj <- RunUMAP(seurat_obj, dims = 1:n_dims)
cat(sprintf("聚类完成: %d 个cluster\n", length(unique(Idents(seurat_obj)))))
# ---- 7. UMAP 图 ----
p_umap <- DimPlot(seurat_obj,
reduction = "umap",
label = TRUE,
label.size = 5,
pt.size = 0.5) +
ggtitle("C2 样本 UMAP") +
theme(plot.title = element_text(hjust = 0.5, size = 16, face = "bold"))
ggsave("C:/Users/BMbio/Desktop/C2/UMAP_clusters.png", p_umap,
width = 8, height = 7, dpi = 200)
cat("UMAP图已保存: UMAP_clusters.png\n")
# ---- 8. 按细胞数量统计 ----
cluster_counts <- table(Idents(seurat_obj))
cat("\n各Cluster细胞数量:\n")
print(cluster_counts)
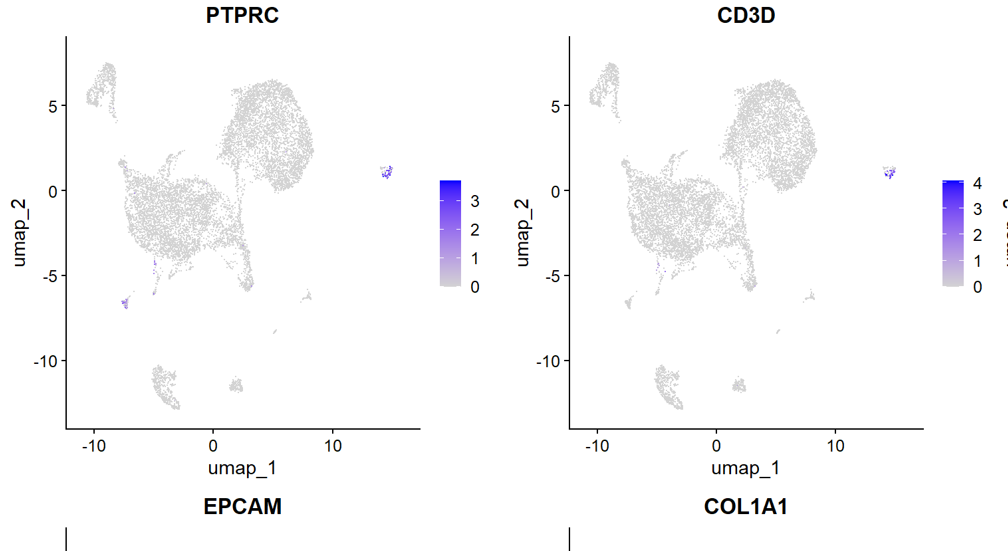
# ---- 9. 特征基因 UMAP (可选) ----
# 常见marker基因
markers_to_plot <- c("PTPRC", "CD3D", "CD14", "EPCAM", "COL1A1")
markers_present <- markers_to_plot[markers_to_plot %in% rownames(seurat_obj)]
if (length(markers_present) > 0) {
p_feat <- FeaturePlot(seurat_obj,
features = markers_present,
ncol = min(3, length(markers_present)))
ggsave("C:/Users/BMbio/Desktop/C2/UMAP_markers.png", p_feat,
width = 5 * min(3, length(markers_present)), height = 5 * ceiling(length(markers_present)/3),
dpi = 150)
cat("Marker基因UMAP已保存: UMAP_markers.png\n")
}
# ---- 10. 保存对象 ----
saveRDS(seurat_obj, "C:/Users/BMbio/Desktop/C2/C2_seurat.rds")
cat("\n分析完成! Seurat对象已保存: C2_seurat.rds\n")
cat("输出文件:\n")
cat(" - QC_violin.png\n")
cat(" - ElbowPlot.png\n")
cat(" - UMAP_clusters.png\n")
if (length(markers_present) > 0) cat(" - UMAP_markers.png\n")如果拿到这些图不知道如何去进行分析,可以询问百沐一下
下面用小提琴质控图为例子进行演示:
百沐一下的回答不要太具体,生信小白也一下就能看得懂:
一、这张图是什么?
这是单细胞分析流程里最常见的QC图之一,通常来自 Seurat 等工具。
横坐标是样本/分组(这里只有 C2 ),纵坐标是对应指标数值。
小提琴图的宽度表示该数值范围内细胞的密度,越宽说明落在该范围的细胞越多。
二、三个指标分别代表什么?
1)nFeature_RNA
表示每个细胞检测到的基因数(number of detected genes)。
-
数值太低:
常提示细胞质量差、RNA降解、捕获效率低,或者其实是空液滴/背景。
-
数值太高:
可能提示doublet/multiplet(一个液滴里混进了两个或多个细胞),尤其当同时伴随 nCount_RNA 很高时更值得警惕。
从图上看:
-
主体分布大约在 1000--2000 左右;
-
有少量细胞上延到 3000--4000+。
解释:
-
主体群体说明大多数细胞能检测到较合理数量的基因;
-
上方长尾提示少数细胞基因数异常偏高,可能是高转录活性细胞,也可能是双细胞。
2)nCount_RNA
表示每个细胞捕获到的总转录本计数/UMI总数。
-
太低:
说明测序深度不足或细胞质量差;
-
太高:
常见于细胞体积较大、转录活跃细胞,或者doublet。
从图上看:
-
主体大约集中在 2000--6000 左右;
-
向上有明显长尾,可到 10000以上甚至接近20000。
解释:
-
大部分细胞捕获量中等,比较常见;
-
高表达长尾细胞需要结合 nFeatureRNA 一起看:如果 nCount RNA 和 nFeature_RNA 同时很高**,要特别怀疑 doublet。**
3)percent.mt
表示线粒体基因转录本占总转录本的比例。
-
这个指标是判断细胞是否受损的重要QC参数;
-
线粒体比例高通常意味着:
-
细胞破裂/凋亡;
-
RNA泄漏;
-
低质量细胞;
-
某些组织本身线粒体含量高,需要具体分析。
-
从图上看:
-
大多数细胞集中在 3%--6% 左右;
-
有一条明显上尾,少数细胞可达到 10%--20% 以上,极个别更高。
解释:
-
主体分布比较理想,说明大部分细胞状态尚可;
-
但存在少量高线粒体比例细胞,这部分通常是QC中过滤的重点对象。
由此看来,百沐可以完全可以一站式解决你的科研问题!
从作图到数据分析,都可以搞定。此外,我们将持续挖掘生物医学人最需要的功能(论文写作、标书辅助写作...),欢迎持续关注,提升你的科研效率!