摘要
碳水化合物活性酶 (Carbohydrate-active enzymes, CAZymes)在糖类消化中至关重要,但目前缺乏用于CAZymes谱分析和解释宏基因组数据中底物偏好的工具。为此,研究人员开发了名为Cayman (Carbohydrate Active Enzymes Profiling of Metagenomes)的CAZymes分析工具,以及一个用于基因组或鸟枪法宏基因组数据集的层级底物注释方案。利用这些工具,研究人员系统性地调查了人类肠道微生物(n=107,683个基因组)中的CAZymes,并鉴定出几种推定的粘蛋白利用细菌,包括Hungatella 和Eisenbergiella 属的物种,这些发现得到了实验验证。研究人员比较了高收入国家(High-income settings, HIS)与低收入和中等收入国家(Low- and middle-income settings, LMIS)的肠道宏基因组(n=3,960)中的CAZymes,发现LMIS宏基因组中富含纤维降解CAZymes,而CAZymes的丰富度在HIS宏基因组中普遍更高。进一步分析(n=1,998)表明,结直肠癌(Colorectal cancer, CRC)患者的宏基因组中靶向纤维的CAZymes减少,而靶向糖胺聚糖(Glycosaminoglycans, GAGs)的CAZymes增加。最后,研究人员通过CAZymes结构域的基因组共定位推断了CAZymes的底物。Cayman具有广泛适用性,可从https://github.com/zellerlab/cayman免费获取。
背景
碳水化合物活性酶 (CAZymes)作用于糖类和糖缀合物,是塑造微生物群落代谢能力的关键因素。人类肠道微生物群能够利用宿主无法消化的、来自饮食的多种(复杂)碳水化合物,因为人类基因组仅编码17种分解代谢CAZymes。虽然肠道微生物在基因组CAZymes库和偏好底物方面差异很大,但许多肠道微生物谱系拥有专门降解纤维的CAZymes。肠道微生物碳水化合物代谢对宿主健康至关重要。膳食纤维(Dietary fibre, DF)可被细菌发酵为短链脂肪酸(Short-chain fatty acids, SCFAs),从而促进上皮屏障完整性和肠道健康。相反,缺乏纤维的饮食可能导致粘液层被侵蚀,因为细菌会转向利用粘液,这可能削弱屏障功能。屏障完整性受损是炎症性肠病和结直肠癌的标志,在这两种疾病中,分别富集了参与降解宿主细胞外基质和粘液的肠道微生物CAZymes。鉴于肠道微生物组作为饮食对宿主健康影响的中介核心作用,理解典型的高收入国家生活方式(通常被称为"西方化")如何塑造CAZymes库,可以为疾病过程提供重要见解。先前的研究揭示了HIS和LMIS人群之间人类肠道微生物组组成的巨大差异,并证实了特定微生物及其功能(包括CAZymes)在"西方化"过程中"消失"的假说。尽管将生活方式二元分类为HIS和LMIS是一种过度简化,但这种对比揭示了关键微生物组功能的变化,例如HIS个体中促进粘蛋白利用的CAZymes相对增加。尽管CAZymes对微生物群落及其宿主都至关重要,但现有研究仅限于少数分离株基因组,并未充分利用当代(宏)基因组数据资源。
宏基因组驱动的CAZymes研究相对缺乏的一个根本原因是缺乏可扩展且易于使用的生物信息学工具:尽管有CAZy数据库(http://www.cazy.org)提供手动管理的CAZymes知识中心,以及用于自动CAZymes注释的计算框架(dbCAN),但人类肠道宏基因组中微生物CAZymes的界定和量化通常由于缺乏开源软件而临时进行。虽然CAZymes已被分为糖苷水解酶(Glycoside Hydrolases, GHs)、多糖裂解酶(Polysaccharide Lyases, PLs)、碳水化合物酯酶(Carbohydrate Esterases, CEs)、糖基转移酶(Glycosyl Transferases, GTs)和碳水化合物结合模块(Carbohydrate-Binding Modules, CBMs;我们将其包含在CAZy催化活性类别中),但底物信息更难获取。尽管底物信息已在CAZy数据库和多个研究小组的整理工作中收集,但可能存在不一致的分类。
- Ducarmon, Q. R., Karcher, N., Giri, S., Tytgat, H. L. P., Delannoy-Bruno, O., Pekel, S., Springer, F., Worz, P., Schudoma, C., Typas, A., & Zeller, G. (2026). Cayman enables large-scale analysis of gut microbiome carbohydrate-active enzyme repertoires. Nature Microbiology. https://doi.org/10.1038/s41564-026-02318-2
- 期刊:Nature Microbiology (IF=19.4)
- 发表时间:2026年4月24日(在线发表)
为此,研究人员开发了一个易于使用且免费提供的生物信息学工具(https://github.com/zellerlab/cayman),用于从鸟枪法宏基因组数据中识别和量化(肠道)微生物群落中CAZymes的丰度。此外,研究人员还提供了一个底物注释方案,通过将CAZymes家族分组为具有生物学意义的底物类别,促进对所得CAZymes谱的解释,这些类别是从CAZy数据库和科学文献中手动整理的。研究人员将这些工具应用于大型肠道细菌基因组集合和宏基因组数据集,展示了具有特定底物利用模式(例如,粘蛋白利用)的细菌物种,以及底物利用如何因宿主生活方式和健康状态而异。
方法
开发与整理CAZymes底物方案
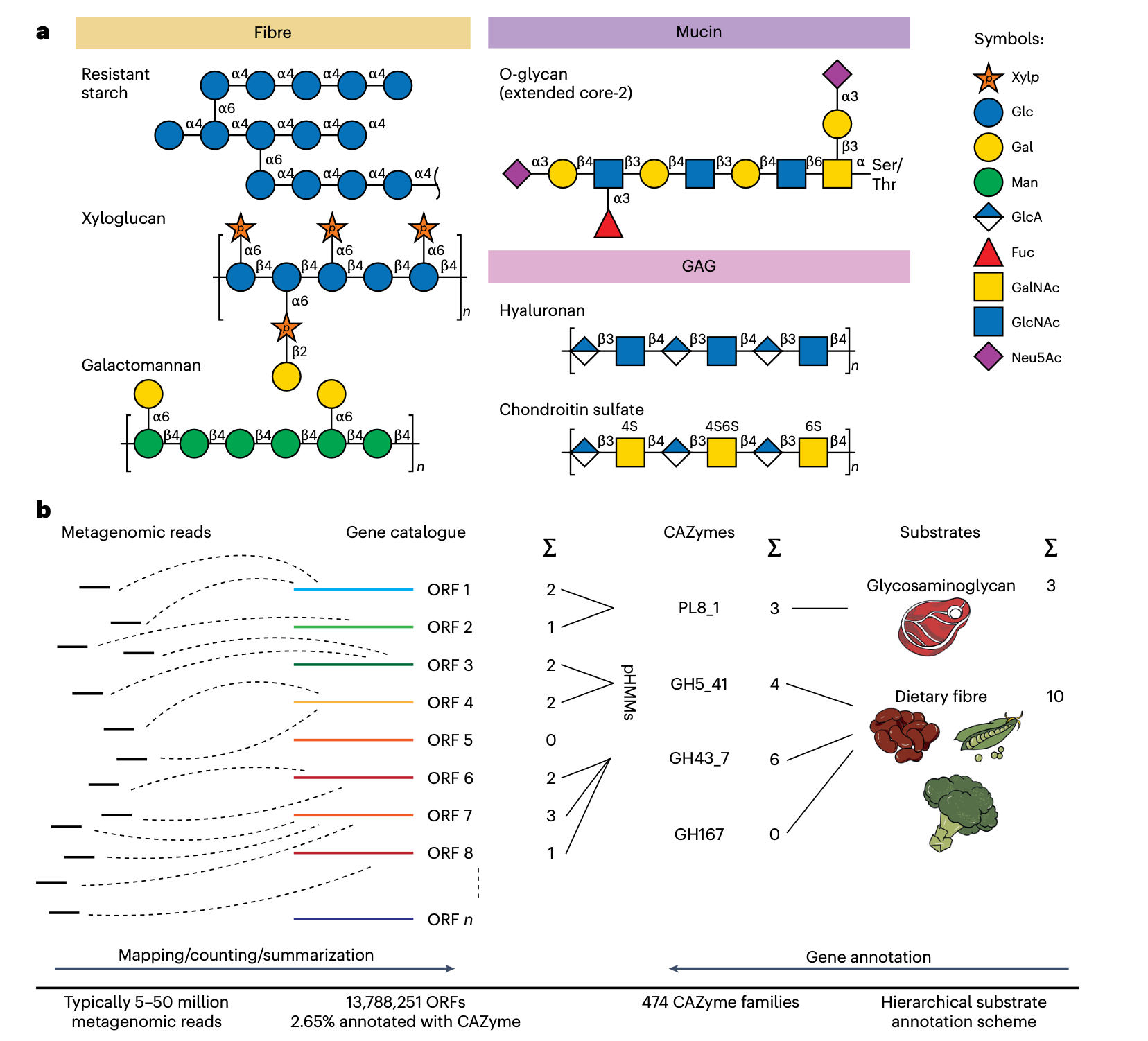
研究人员开发了一个全面的层级CAZymes底物注释方案,旨在克服先前工作通常不完整且仅关注GHs的局限性。三位作者从CAZy数据库(http://www.cazy.org)和科学文献中独立收集了dbCAN2版本9中所有CAZymes类别的底物信息。收集了共识底物注释,并比较了Cayman和dbCAN3之间的一致性。
研究人员收集了多个层次的底物注释类别,主要遵循先前对糖类分类的建议。底物注释有助于在不同粒度级别上解释下游统计分析。例如,CBM2结合纤维素、半纤维素和几丁质(注释级别3),这些是属于膳食纤维(注释级别1)的结构性来源的非淀粉多糖(Non-starch polysaccharides, NSPs;注释级别2)。需要注意的是,与CBM2类似,许多CAZymes家族是多特异性的,这也反映在研究人员每个CAZymes家族可能有多个底物类别的注释中。
Cayman:一种宏基因组CAZymes分析工具
为了能够常规量化宏基因组中的CAZymes,研究人员开发了一个名为Cayman的计算CAZymes分析工具。其功能在以下方面与dbCAN2/3和其他CAZy相关计算流程不同:Cayman不是直接在(短)宏基因组测序读段中筛选CAZymes基因片段,而是首先将读段映射到一个代表微生物基因近乎完整集合的基因目录。在这里,为了分析人类粪便宏基因组,研究人员依赖于一个整合的、非冗余的人类肠道基因目录,在其中使用重新校准的轮廓隐马尔可夫模型(profile Hidden Markov Models, pHMMs)注释了CAZymes。通过基因目录映射进行功能分析,与直接注释读段本身相比,提供了更快、更准确的基因丰度估计。Cayman最终从映射读段的计数中计算长度和测序深度归一化的CAZymes家族丰度,这些丰度可以在给定宏基因组中进一步汇总为更高级别的类别。
Cayman广泛适用于来自其他环境的(宏)基因组数据,尽管这些环境缺乏专门的基因目录(研究人员提供了构建此类目录的说明,详见https://github.com/zellerlab/cayman)。
输入: 宏基因组测序数据
基因目录映射
使用pHMMs注释CAZymes
计算CAZymes家族丰度
应用层级底物注释方案
生成CAZymes谱
统计分析
比较不同人群/疾病状态
识别差异丰度CAZymes
推断底物偏好
输出: CAZymes丰度谱、
底物富集分析、差异表达结果
结果
人类肠道微生物CAZymes库的基因组探索
由于宏基因组组装的进步,人类肠道微生物组的基因组资源大幅增长。为了提供人类肠道微生物CAZymes库的最新视图,研究人员重新注释了107,683个高质量宏基因组组装基因组(Metagenome-Assembled Genomes, MAGs)和分离株基因组中的基因。分析证实,糖类代谢的基因组潜力在分类学上差异很大。除了确认拟杆菌门(Bacteroidetes)的关键属(最突出的是拟杆菌属和副拟杆菌属)拥有广泛的CAZymes库外,研究人员还发现厚壁菌门(Firmicutes)中的几个属也富含CAZymes。其中,Hungatella 和Eisenbergiella 尤为突出,分别拥有78个家族的224个基因和106个家族的404个基因。
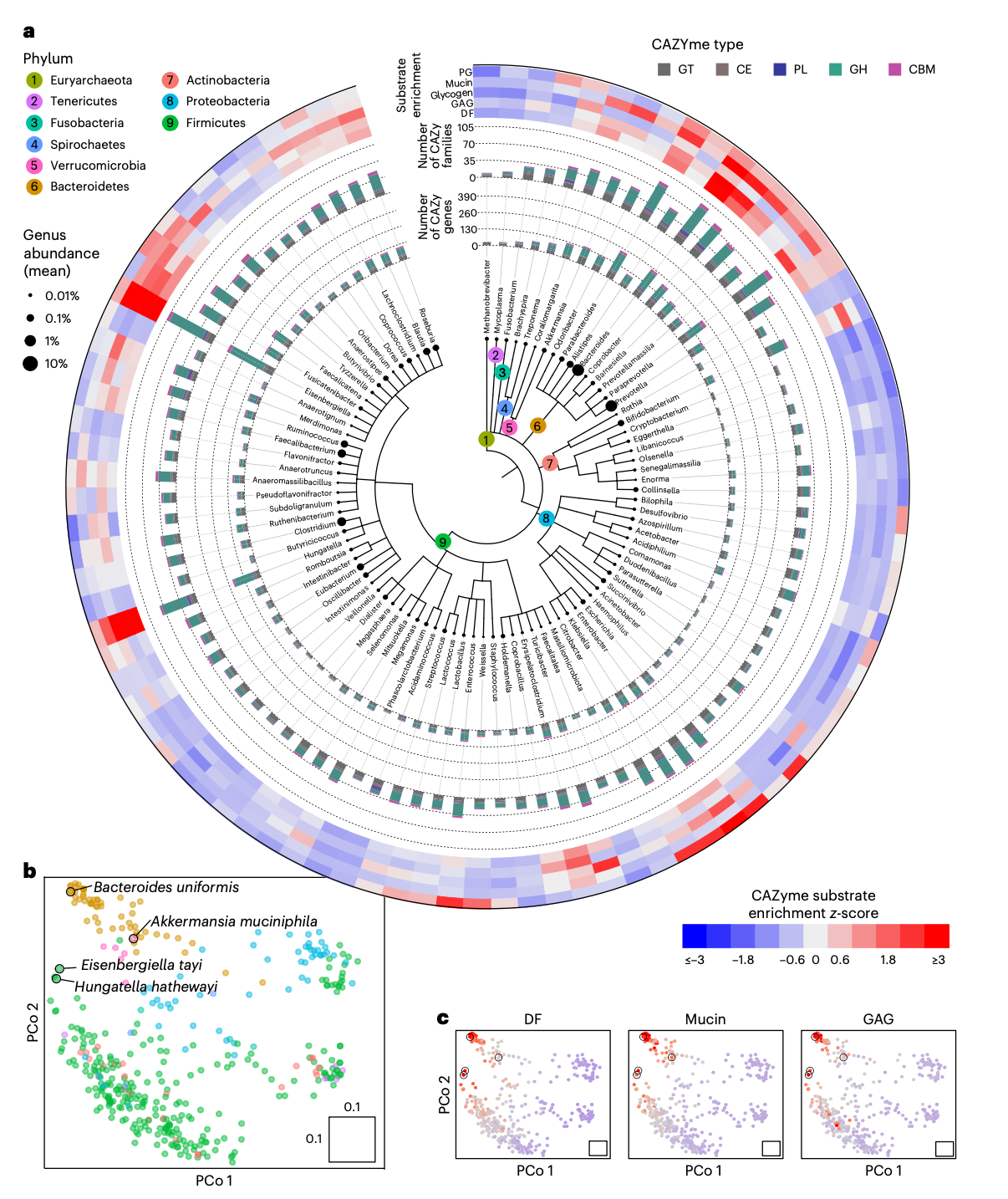
研究人员进一步基于CAZy家族存在与否的成对相似性,使用排序方法比较了肠道微生物类群的整体CAZymes库。来自同一门的物种倾向于根据其CAZymes库进行聚类。此外,研究人员利用底物注释计算了五个主要底物类别的富集分数:肽聚糖(Peptidoglycan, PG)、粘蛋白(Mucin)、糖原(Glycogen)、糖胺聚糖(GAG)和膳食纤维(DF)。该分析证实了已知的粘蛋白利用菌,如Akkermansia、Alistipes和Bacteroides,拥有许多不同的靶向粘蛋白的CAZymes基因。特征描述较少的Barnesiellaceae科包括Barnesiella和Coprobacter两个属,其成员也富含靶向粘蛋白的CAZymes。
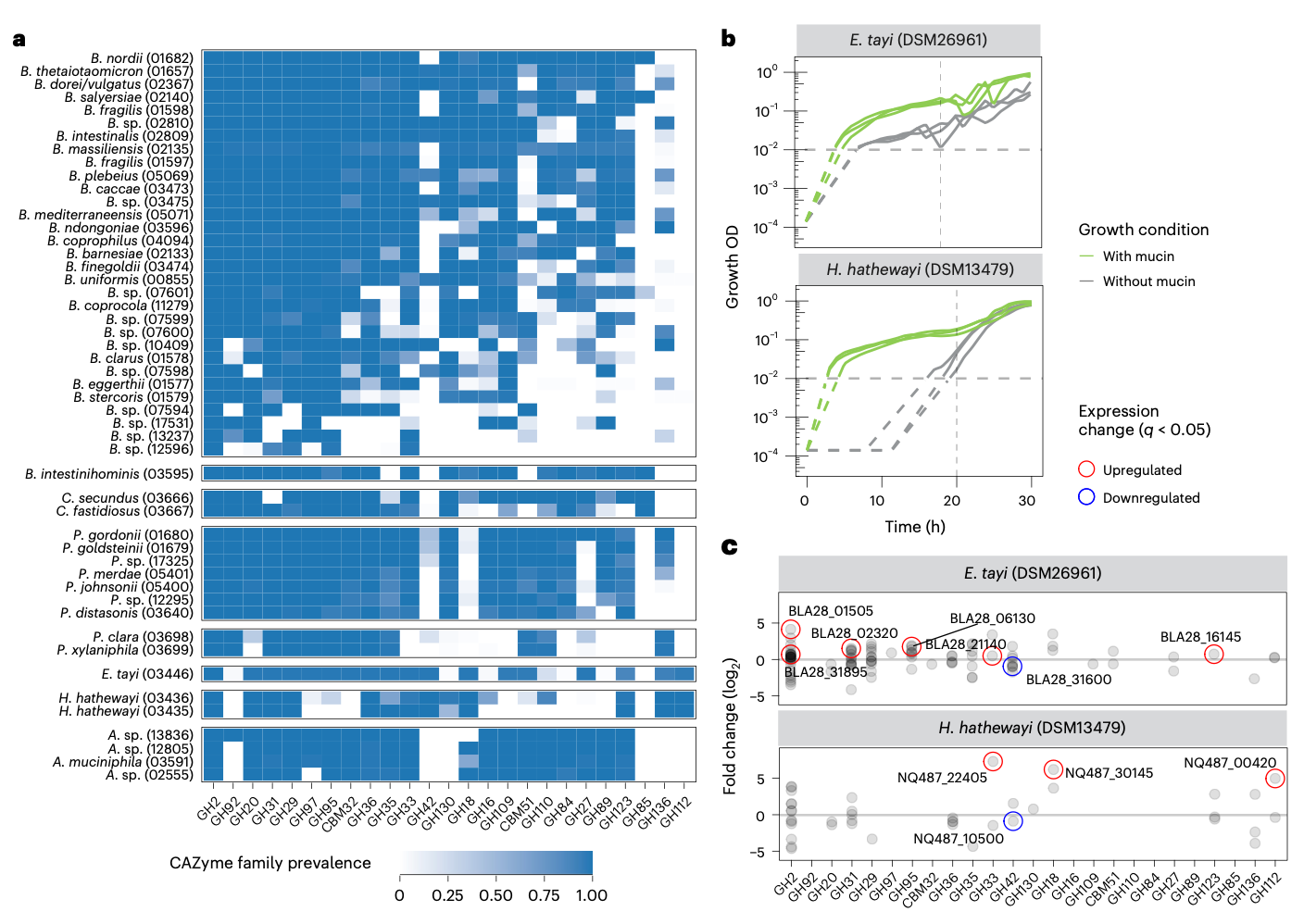
为了在物种分辨率上解析靶向粘蛋白的CAZymes库,研究人员调查了在粘蛋白底物上富集最强的八个属中的相关CAZymes家族。虽然该分析揭示了拟杆菌属内CAZymes库存在相当大的物种水平变异,但其他属似乎异质性较低,部分原因是它们在人类肠道中的物种成员数量要少得多。虽然B. intestinihominis已被描述为粘蛋白专家,可以专门在粘蛋白O型聚糖上生长,但据研究人员所知,Coprobacter的两个物种尚未有此类报道,而研究结果表明它们是高效的粘蛋白利用菌。最后,研究人员发现Hungatella和Eisenbergiella基因组在靶向粘蛋白的CAZymes上强烈富集,H. hathewayi和E. tayi的整体CAZymes库与已知的粘蛋白利用菌相似。
为了获得E. tayi和H. hathewayi潜在粘蛋白代谢能力的进一步证据,研究人员使用SulfAtlas工具筛选了它们的基因组中靶向结肠粘蛋白的硫酸酯酶(S1_4, S1_11, S1_15, S1_16和S1_20)。这揭示了在E. tayi和H. hathewayi的基因组中存在大多数已知的靶向结肠粘蛋白的硫酸酯酶。总之,该分析系统地概述了人类肠道细菌属和物种的糖类相关代谢潜力。
H. hathewayi和E. tayi粘蛋白利用潜能的实验验证
为了通过实验测试H. hathewayi、E. tayi和Coprobacter secundus是否能够利用粘蛋白,研究人员在含有和不含粘蛋白的培养基中培养了这些物种。研究人员观察到,在存在粘蛋白的情况下,所有三个物种以及阳性对照Akkermansia muciniphila在Wilkins-Chalgren厌氧菌(WCA)培养基中生长更好(生长更快、生长速率增加和/或产量更高)。为了确定哪些CAZymes可能促进粘蛋白代谢,研究人员评估了E. tayi和H. hathewayi(这两个物种在WCA中生长到更高产量)在培养物达到平台期时间点的转录活性。在进行RNA测序(RNA-seq)后,研究人员计算了存在和不存在粘蛋白条件下的差异丰度转录本。该分析揭示了两个物种中几种上调的CAZymes,指出了可能参与粘蛋白代谢的蛋白质。这些结果表明,H. hathewayi和E. tayi在富含粘蛋白的培养基中生长更好,表明它们可以代谢利用粘蛋白,从而支持了研究人员的计算预测。
HIS与LMIS肠道宏基因组的荟萃分析
从单个肠道微生物转向群落CAZymes库,研究人员通过将Cayman应用于公开可用的HIS和LMIS肠道宏基因组(n=3,166来自HIS个体,n=794来自LMIS个体),研究了这些库在人群中的差异。CAZy谱的主坐标分析显示HIS和LMIS微生物组之间存在明显分离。
先前的小规模研究描述,与HIS宏基因组相比,来自LMIS国家的宏基因组具有更高的CAZymes多样性,这种现象主要归因于HIS饮食中膳食纤维含量低。与此形成鲜明对比的是,研究人员发现与LMIS人群相比,HIS人群中独特CAZymes的数量持续更高,即使只考虑参与膳食纤维代谢的CAZymes也是如此。虽然研究人员注意到与LMIS宏基因组相比,HIS宏基因组的读段映射率更高,但对具有相似读段映射率的HIS和LMIS样本的额外比较使这种解释不太可能成立。
接下来,研究人员利用底物注释方案比较了HIS与LMIS的CAZymes库。研究人员证实了先前的发现,即与LMIS宏基因组相比,HIS宏基因组中靶向粘蛋白与靶向膳食纤维的CAZymes丰度比更高。类似地,与靶向膳食纤维的CAZymes相比,HIS宏基因组中靶向GAGs(动物来源的聚糖;肠道GAGs的来源包括脱落的上皮细胞和饮食摄入)的CAZymes丰度比更高。这些结果与HIS饮食通常含有大量动物性产品且纤维含量低,而许多LMIS饮食更多以植物为基础且富含纤维的情况一致,尽管这些饮食内部存在很大差异。最后,研究人员在底物层级更精细的级别上进行了底物富集分析,揭示了HIS宏基因组中参与糖蛋白代谢的CAZymes富集,有趣的是,在各种非淀粉多糖(如果胶和树胶)中也富集。鉴于果胶和树胶是加工食品中常见的添加剂,这些富集可能源于HIS人群对加工食品的摄入量更高。
总之,HIS宏基因组在降解宿主和动物来源聚糖以及加工食品中常见的非淀粉多糖的肠道微生物代谢潜力方面,其丰富度和丰度都有所增加。
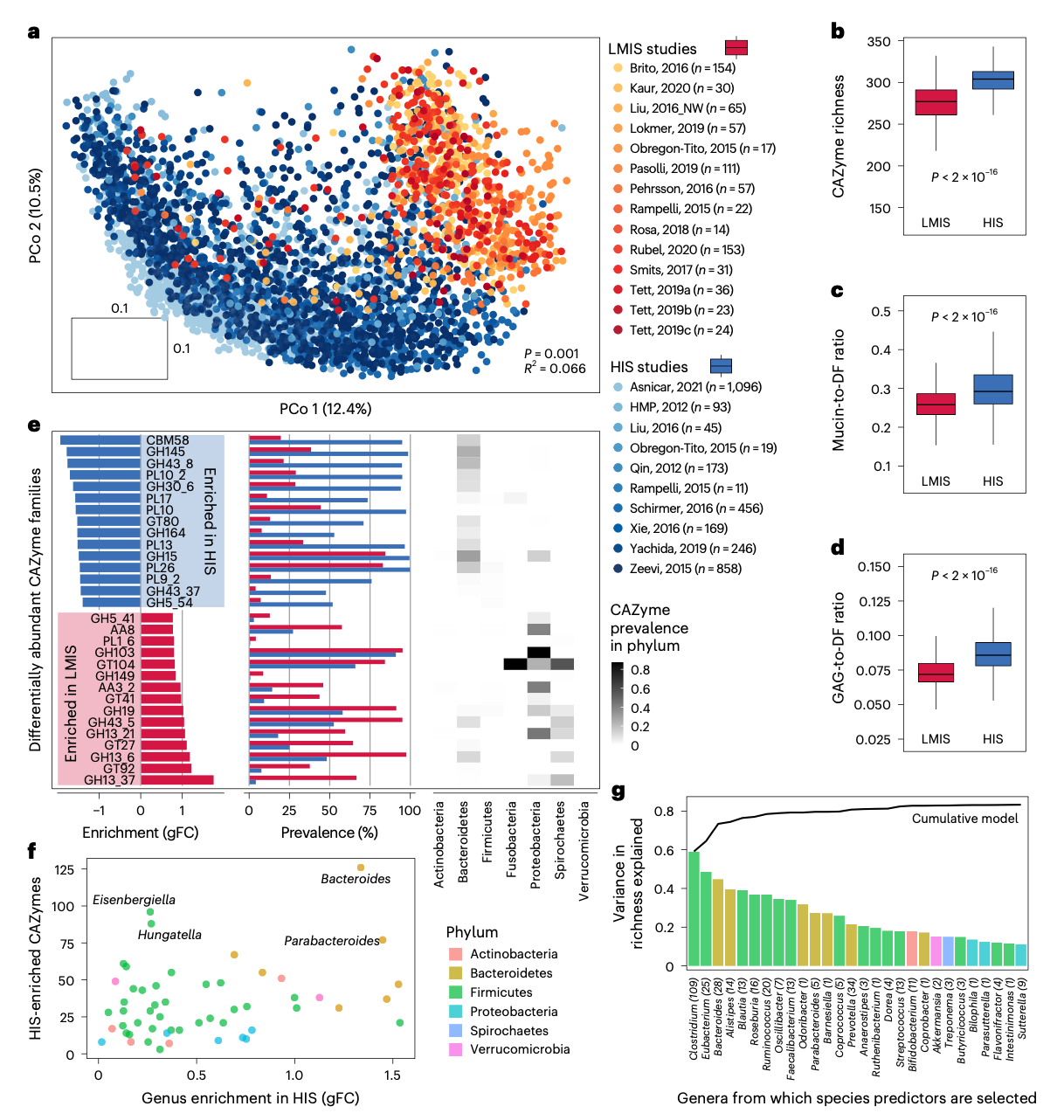
然后,研究人员使用线性模型对每个CAZymes家族进行HIS和LMIS之间的差异丰度测试。该分析表明许多CAZymes在丰度和流行率上存在显著差异。在最富集于LMIS的CAZymes中,研究人员观察到四个靶向抗性淀粉的家族(GH13_37、GH13_6、GH13_21和GH13_42),这与LMIS人群普遍摄入更多全谷物和豆类一致。为了将差异家族归因于其可能的分类学起源,研究人员比较了不同门中CAZymes的流行率。这揭示了许多富集于LMIS的CAZymes在变形菌门(Proteobacteria)和螺旋体门(Spirochaetes)中很普遍。后一个门被标记为"消失"的分类群,在LMIS中常见但在HIS微生物组中丢失。相比之下,最富集于HIS的CAZymes------CBM58,是SusG蛋白的一部分,该蛋白对拟杆菌属物种的淀粉利用至关重要,而拟杆菌属在HIS个体中往往丰度更高。
为了评估拟杆菌属的扩张是否可以解释HIS人群中CAZymes丰富度的增加,研究人员询问每个(富集于HIS的)细菌属可以为群落库贡献多少富集于HIS的CAZymes。该分析确定拟杆菌属拥有最多数量的富集于HIS的CAZymes。接下来,研究人员调查了少数关键类群的基因组CAZymes库在多大程度上可以预测跨人群的整个群落的CAZymes丰富度。为此,研究人员用逐步增加的分类预测因子集构建了基于属丰度的CAZymes丰富度线性回归模型。CAZymes丰富度可以通过几个属很好地预测。然而,虽然拟杆菌属是这些预测因子之一,但真杆菌属(Eubacterium)和梭菌属(Clostridium)甚至更能预测群落CAZymes丰富度。这些结果表明,虽然富集于HIS的拟杆菌属在基因组上拥有最丰富的与HIS相关的CAZymes,但其丰度变化本身并不能解释群落的CAZymes含量。相反,几个关键类群共同似乎能更好地解释跨人群的个体间CAZymes丰富度差异。
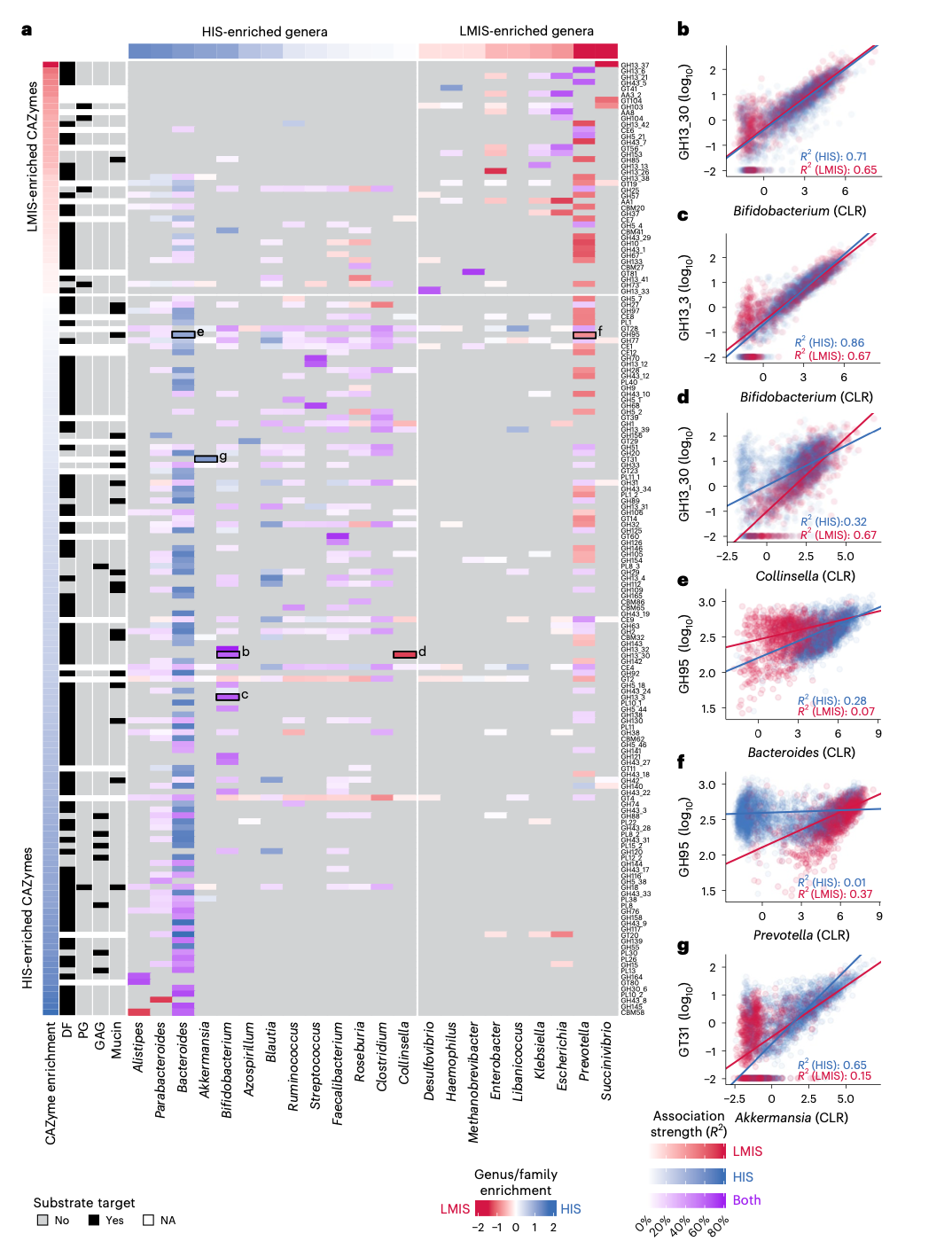
为了将分类组成与群落CAZymes库联系起来,研究人员构建了线性回归模型,根据每个肠道微生物属内的物种丰度谱来预测每个CAZymes家族的丰度。研究人员分别针对HIS和LMIS人群训练这些模型以比较关联。研究人员发现了分类群丰度和CAZymes丰度之间的强成对关联,并且正如预期的那样,观察到高丰度属(如拟杆菌属和普雷沃菌属)对CAZymes丰度的贡献通常超过稀有、低丰度属。然而,研究人员注意到这一趋势的例外情况,例如双歧杆菌属(Bifidobacterium)与GH13_30和GH13_3。GH13_30在双歧杆菌属物种中已有很好的表征,因此与双歧杆菌属丰度的强关联似乎是合理的。相比之下,GH13_3在任何肠道微生物中尚未有实验表征,但分析显示GH13_3丰度可以从双歧杆菌属物种的丰度很好地预测,表明该属是HIS和LMIS人类肠道微生物组中GH13_3家族的唯一贡献者。其次,研究人员观察到,尽管类群丰度相似,但某些关联在HIS和LMIS个体之间存在差异:例如,Collinsella在LMIS个体中强烈预测GH13_30丰度,但在HIS个体中则不然。Collinsella丰度没有显著差异这一事实表明,其他类群在HIS而非LMIS个体中贡献了这个CAZymes库。一个生态上不同的例子是GH95(编码α-岩藻糖苷酶):在HIS个体中,这个CAZymes家族与拟杆菌属强烈相关,但与普雷沃菌属无关,而在LMIS个体中观察到相反的模式。这表明在HIS中,拟杆菌属接管了原本由普雷沃菌属提供的功能。这个例子特别有趣,因为普雷沃菌属通常不被视为粘蛋白利用菌。此外,尽管该家族已在许多肠道微生物中表征,但尚未有来自普雷沃菌属的酶被实验鉴定为GH95。作为最后一个例子,Akkermansia在HIS个体中高度预测GT31,但在LMIS个体中则不然,表明Akkermansia是HIS个体中该CAZymes家族的主要携带者,但在LMIS个体中则不是。相比之下,在LMIS个体中,单细胞寄生虫Blastocystis(其基因组也编码GT31)与GT31丰度的关联比Akkermansia更强。
CRC病例对照研究的荟萃分析
HIS生活方式与许多常见疾病相关,包括结直肠癌。CRC患者与对照组之间肠道微生物组成的差异已得到很好理解,并且个别研究也报告了它们CAZymes库的差异。在此,研究人员通过将Cayman应用于来自四大洲十个不同国家的1,998个宏基因组(n=968名CRC患者和n=1,030名对照)的荟萃分析,提供了肠道CAZymes库的详细评估。
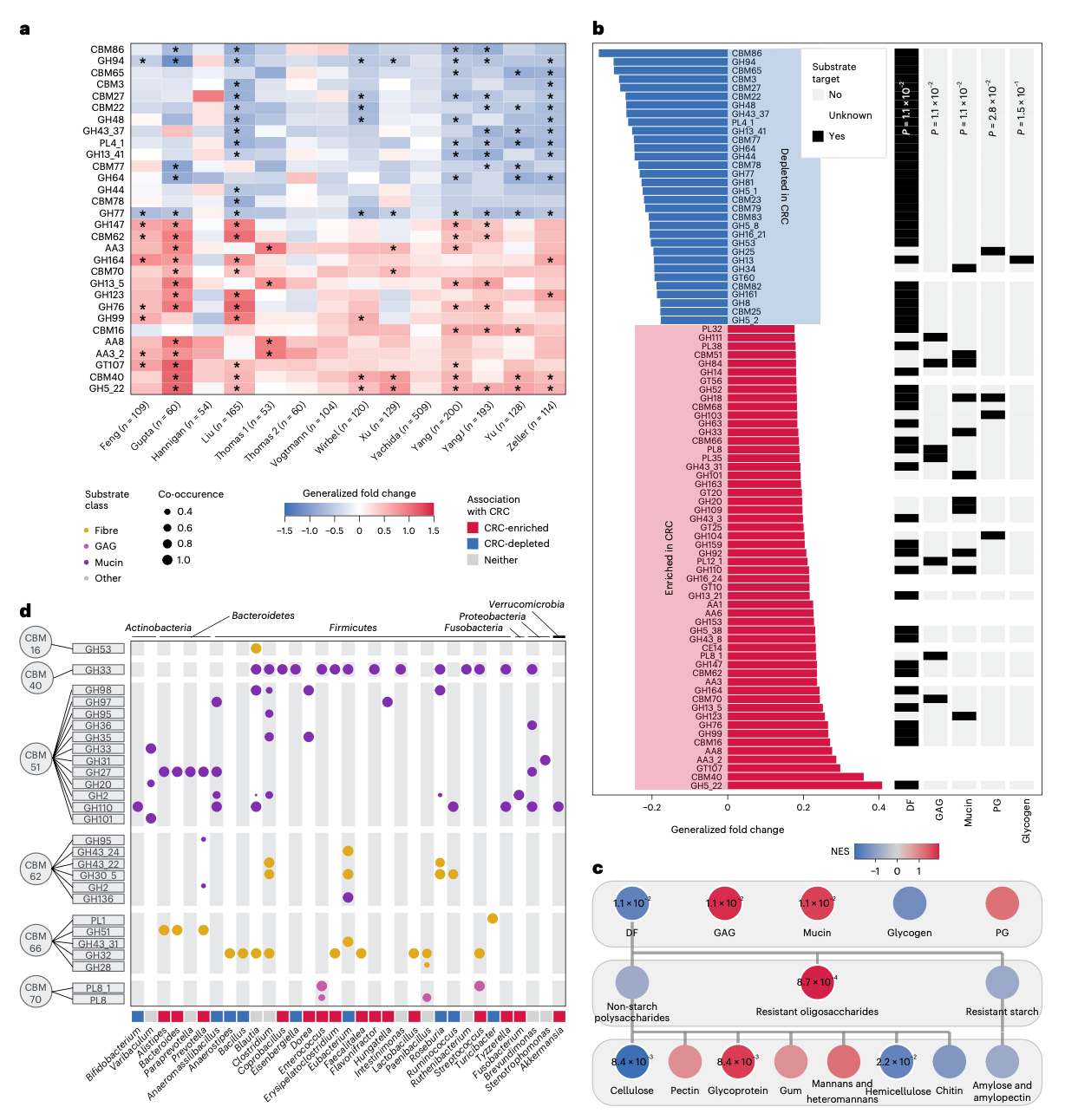
研究人员首先计算了每项研究中差异丰度的CAZymes家族,揭示了跨研究中CRC患者CAZymes富集和耗竭的广泛一致性;然而,与先前的荟萃分析一致,几个数据集在单独评估时并未显示显著差异。
为了进行差异丰度CAZymes家族的荟萃分析,同时考虑研究异质性,研究人员拟合了线性混合模型,以CAZymes谱预测癌症状态,并将研究作为随机效应。在459个CAZymes中,有282个在5%错误发现率(False Discovery Rate, FDR)下显著。接下来,研究人员旨在通过利用底物方案进行基因集富集分析(Gene Set Enrichment Analysis, GSEA)来识别碳水化合物底物偏好的潜在群落范围变化。研究人员发现CRC宏基因组中具有膳食纤维来源底物的CAZymes家族显著耗竭,同时还注意到GAG和粘蛋白底物的富集。粘蛋白与膳食纤维以及GAG与膳食纤维的比率进一步证实了这一发现,两者在CRC宏基因组中均增加。在不同粒度级别上的底物富集分析揭示了纤维素和半纤维素底物等的显著耗竭。总之,这些结果显示CRC宏基因组中宿主和动物聚糖底物明显富集,而膳食纤维来源的底物则耗竭。这些发现与早期报告以及CRC的饮食流行病学风险因素一致,并且可能是饮食差异或肠道微生物组对CRC中宿主聚糖可用性增加的生态适应的结果。
为了更多地了解富集于CRC的CBMs的底物,研究人员调查了这些CBMs是否与特定的CAZymes催化结构域共定位,从而推断其底物。分析揭示了几个CRC富集的CBMs与靶向粘蛋白和GAGs的CAZymes结构域共定位,支持了CRC微生物组转向利用宿主来源聚糖的假设。
讨论
本研究开发的Cayman工具及其配套的层级底物注释方案,为大规模、系统性地分析宏基因组中的CAZymes提供了强大且用户友好的解决方案。通过将读段映射与预注释的基因目录相结合,Cayman克服了传统方法在准确性和计算效率上的局限,使得对复杂微生物群落中碳水化合物代谢潜能的深入探索成为可能。
对超过10万个肠道微生物基因组的分析,不仅更新了我们对关键菌属CAZymes库的认识,更重要的是发现了Hungatella和Eisenbergiella等新的潜在粘蛋白利用菌。实验验证进一步支持了这些计算预测,表明这些菌株在粘蛋白存在时生长增强,并且相关CAZymes基因表达上调。这扩展了我们对肠道微生物粘蛋白代谢网络的理解,并提示这些菌株可能在肠道屏障功能中扮演尚未被充分认识的角色。
HIS与LMIS人群的宏基因组比较揭示了令人意外的模式:尽管LMIS饮食通常富含纤维,但HIS个体的肠道微生物组却拥有更高的CAZymes基因丰富度。这一发现挑战了简单将CAZymes多样性与纤维摄入量直接关联的观点。相反,它表明HIS生活方式可能通过其他途径(如加工食品添加剂、动物性产品摄入)塑造了独特的CAZymes库,其中包含更多靶向宿主来源聚糖(粘蛋白、GAGs)和食品添加剂(如果胶、树胶)的酶。分类学关联分析进一步揭示,HIS中CAZymes库的扩张并非仅由单一优势菌属(如拟杆菌属)驱动,而是多个关键类群共同作用的结果,并且不同类群对特定CAZymes的贡献在HIS和LMIS人群间存在生态位替代现象。
在CRC的荟萃分析中,观察到的CAZymes谱变化------即纤维降解酶减少,而粘蛋白和GAG降解酶增加------与疾病状态下肠道微环境的改变相一致。粘液层变薄和宿主组织破坏可能为微生物提供了更多的宿主聚糖来源,从而选择了具有相应降解能力的菌株。这种CAZymes谱的转变不仅可能是CRC的结果,也可能通过影响屏障功能、炎症和微生物代谢产物而参与疾病进程。
结论
Cayman工具的开发及其在大型数据集上的应用,极大地推进了我们对人类肠道微生物组碳水化合物代谢潜能的理解。该研究系统性地描绘了肠道微生物CAZymes库的多样性,揭示了生活方式(HIS vs. LMIS)和疾病状态(CRC)对微生物碳水化合物代谢能力的深刻影响。
主要结论包括:
- 工具创新:Cayman提供了一个高效、准确的框架,用于从宏基因组数据中分析CAZymes,其层级底物注释方案显著提升了对CAZymes功能意义的解读能力。
- 新菌株发现:研究鉴定出Hungatella hathewayi和Eisenbergiella tayi等新的潜在粘蛋白利用细菌,并通过实验验证了其代谢能力,拓宽了已知的粘蛋白降解菌名录。
- 生活方式影响:HIS人群肠道微生物组拥有更高的CAZymes基因丰富度,但底物偏好转向宿主来源聚糖和加工食品添加剂,反映了饮食西化对微生物功能的复杂重塑。
- 疾病关联:CRC患者的肠道微生物组表现出CAZymes谱的显著改变,即纤维降解能力下降,而宿主聚糖(粘蛋白、GAGs)降解能力增强,这为理解CRC的微生物组机制提供了新线索。
这些发现强调了在菌株和功能水平上解析微生物组的重要性。Cayman作为一个开源工具,将促进未来更多研究探索CAZymes在人类健康与疾病、饮食干预以及生态系统功能中的作用。未来的工作可以进一步将CAZymes谱与详细的饮食记录、宿主表型以及代谢组学数据相结合,以建立更精确的因果关系,并探索基于CAZymes谱的疾病诊断或干预靶点。