The genome of Magnolia biondii Pamp. provides insights into the evolution of Magnoliales and biosynthesis of terpenoids
望春玉兰基因组解析揭示木兰目演化历程与萜类物质生物合成机制

摘要
望春玉兰隶属于木兰科木兰类植物,是兼具系统发育研究价值、经济价值与药用价值的观赏树种,在中国北温带地区被广泛引种栽培。解析望春玉兰基因组序列,有助于厘清木兰类植物系统发育地位争议,加深对木兰属物种特有性状演化规律的认知。
本研究整合 PacBio 单分子实时测序、10X Genomics 及 Hi-C 染色体挂载技术,分别获得约 67 Gb、175 Gb、154 Gb 原始测序数据,成功组装获得望春玉兰染色体水平参考基因组。最终组装基因组大小约 2.22 Gb,Contig N50 长度为 269.11 kb,BUSCO 完整基因占比达 91.90%;其中 89.17% 的基因组序列成功锚定至 19 条染色体,Scaffold N50 长度为 92.86 Mb。
基因组共注释得到 47547 个蛋白编码基因,占基因组全长的 23.47%,重复序列占比高达 66.48%。本研究证实望春玉兰经历过一次全基因组复制事件,该事件发生时间与木兰目和樟目物种分化时期高度相近。
木兰属特有基因及扩张基因家族的功能富集结果显示,这类基因主要参与次生代谢产物生物合成、植物与病原菌互作及环境胁迫应答通路,有助于提升该类植物的生态适合度与环境适应能力。
系统发育基因组学分析表明,木兰类植物与金粟兰科植物互为姊妹群,二者共同构成单子叶植物与双子叶植物演化支的外类群。望春玉兰基因组数据可为林木性状改良、种质资源保护,以及早期被子植物快速辐射演化相关研究提供重要参考依据。
引言
木兰科植物全球现存物种超 300 种,主要分为两大属:仅含 2 个物种的鹅掌楸属,以及其余绝大多数物种所属的木兰属。现存约八成木兰科植物自然分布于东南亚温带与热带区域,剩余物种分布于美洲地带,范围涵盖北美东南部温带区域、中美洲至巴西,形成典型的间断地理分布格局。
木兰属隶属于木兰类植物,木兰类是被子植物中演化分化较早的类群,在被子植物系统发育演化进程中占据核心关键地位。被子植物早期演化分化出无油樟目、睡莲目等基部类群后,中生被子植物五大演化支系(木兰类、金粟兰科、金鱼藻目、单子叶植物、双子叶植物)在不足 500 万年的极短时间内发生快速辐射演化,致使部分中生被子植物类群间的系统发育关系至今仍存在争议。
目前已公开的被子植物基因组数据共 323 套,大多集中于农作物物种,而木兰类植物仅完成 5 个物种基因组测序,分别为胡椒、牛油果、刺果番荔枝、樟及鹅掌楸。依托现有基因组数据开展的系统发育分析,使得木兰类植物的演化地位始终没有统一结论:部分研究结果强力支持木兰类与双子叶植物互为姊妹群 ,该结论与 92 种链形植物、20 种代表性被子植物的转录组系统发育分析结果一致;另有多项研究仅得到较弱证据,认为木兰类是单子叶与双子叶植物共同演化支的姊妹群 ,这一观点也与陆生植物、绿色植物及被子植物质体基因组大规模系统发育分析结论相符。系统发育推演结果高度依赖物种取样范围、基因集选取与分析方法,因此亟需补充更多木兰类物种基因组数据,厘清其与单、双子叶植物之间的演化亲缘关系。
木兰属植物多为雌蕊先熟的异花授粉植物,有效授粉周期极短;同时该属多数物种花粉萌发率、种子萌发率偏低,结实量少,自然种群自我更新能力极差。再加上人为伐木破坏、农业开发导致原生栖息地锐减,近半数木兰属野生物种已处于濒危状态。开展木兰属植物种质资源保护兼具重要经济价值与生态价值。多数木兰属树姿优美、花香馥郁,是极具观赏价值的园林绿化树种,如白玉兰、紫玉兰、广玉兰等;其花器官富含大量萜类物质,树皮中含有丰富酚类活性成分,望春玉兰、厚朴、白玉兰、武当玉兰等诸多物种更是传统药用与美妆原料植物。但目前缺乏木兰属高质量参考基因组,严重制约该属植物种质资源保护与开发利用,完整的基因组信息能够为木兰属分子育种、种质保育及基础科研提供核心数据支撑。
望春玉兰是集观赏、药用与用材价值于一体的木兰属树种,为落叶乔木,在中国北温带地区广泛栽培种植。其花大浓香,可提取芳香精油;玉兰花蕾提取物具备局部刺激、麻醉、抗炎、抑菌、镇痛、降压、抗过敏等多种药理活性。现代植物化学研究已系统鉴定出望春玉兰不同组织中的挥发油、木质素、生物碱等化学成分,其挥发油内含大量萜类物质,以 1,8 - 桉叶素、β- 蒎烯、α- 松油醇、樟脑为主要活性组分,这类萜类化合物均由萜类合成酶基因家族编码蛋白催化合成。
本研究利用 PacBio 长读长测序、10X Genomics 及 Hi-C 染色体挂载技术,完成望春玉兰全基因组测序与组装,组装基因组大小约 2.22 Gb,是目前已发表的基部木兰类植物中基因组规模最大的物种。该基因组数据可为木兰类花器官演化、特有初生与次生代谢物质合成机制研究提供数据基础,也将助力解析被子植物主干谱系快速演化历程,同时推动木兰属植物基因组辅助育种、人工栽培及野生种质保护相关研究。
材料与方法
植物材料、DNA 提取与测序
试验所用材料取自西安植物园内一株树龄 21 年的人工栽培望春玉兰植株,分别采集不同发育阶段的新鲜叶片与花组织;植物凭证标本(编号:Zhang 201801M)保存于深圳仙湖植物园标本馆。
采用改良 CTAB 法提取望春玉兰幼嫩叶片高质量基因组 DNA,利用紫外分光光度计与荧光定量仪分别完成 DNA 纯度与浓度检测。所有基因组测序工作均在深圳华大基因完成,共设置三套测序体系:
- 依据官方实验流程构建插入片段 350~500 bp 的 10X Genomics 测序文库,搭载 BGISEQ-500 平台完成 150 bp 读长测序;使用 SOAPnuke 软件过滤重复序列、低质量碱基占比超 20%、未知碱基 N 占比超 5% 的无效测序 reads。
- 按照 PacBio 20 kb 建库标准构建单分子实时测序文库,于 PacBio RS-II 测序平台开展长片段测序。
- 利用 DpnII 限制性内切酶结合原位连接法构建 Hi-C 文库,对酶切染色质进行生物素标记与原位 DNA 连接,纯化后片段经超声破碎、末端加 A、磁珠富集、接头连接,最终在 BGISEQ-500 平台完成 100 bp 读长测序。
RNA 提取与转录组测序
采集同一株望春玉兰的幼嫩叶片、盛花期花朵,以及减数分裂前、减数分裂后两个发育时期的花蕾样品;使用植物总 RNA 提取试剂盒提取各样品总 RNA,经质检筛选完整性合格的 RNA 样品,构建 cDNA 文库后利用 Illumina HiSeq 2000 平台完成双端 150 bp 转录组测序,最后通过 Trimmomatic 软件去除测序接头、低质量序列及短小片段,获得干净转录组数据。
基因组大小预估
利用 10X Genomics 小片段文库高质量测序数据开展 17-mer 频数统计,借助 k-mer 分布特征结合 GCE 软件,预估望春玉兰基因组全长、重复序列占比与基因组杂合度。
基因组从头组装与染色体构建
分别使用 Canu、Miniasm、Wtdbg、Flye、SMARTdenovo 多款主流组装软件进行基因组初步组装,综合组装基因组总长、Contig 数量、Contig N50、最长序列长度及 BUSCO 基因组完整度评估结果,最终选定 Miniasm 组装结果进行后续纠错与染色体挂载。
首先利用 Racon 软件依托全部 PacBio 长读长序列完成三轮迭代纠错,再使用 Pilon 软件结合 10X Genomics 干净二代数据完成单轮碱基精准校正;随后运用 Juicer 软件将 Hi-C 有效测序 reads 比对至校正后的 Contig 基因组,通过 3D-DNA 软件完成序列纠错、排序、定向排布,初步搭建染色体水平基因组框架,最后借助 Juicebox 可视化工具完成人工微调修正,获得精准染色体水平参考基因组。
|------------------------------------|-----------------------|--------|
| PacBio Assembly (polished) | Hi-C Assembly |
| Total scaffold length (Gb) | | 2.232 |
| Number of scaffolds | | 9510 |
| Scaffold N50 (Mb) | | 92.86 |
| Scaffold N90 (Mb) | | 19.29 |
| Max scaffold length (Mb) | | 168.50 |
| Total contig length (Gb) | 2.22 | |
| Number of contigs | 15,615 | |
| Contig N50 (kb) | 269.114 | |
| Contig N90 (kb) | 60.09 | |
| Max contig length (kb) | 2,134.98 | |
| Complete BUSCOs | 91.90% | 88.50% |
| Complete and single-copy BUSCOs | 87.00% | 85.20% |
| Complete and duplicate BUSCOs | 4.90% | 3.30% |
| Fragmented BUSCOs | 3.00% | 4.40% |
基因组组装质量评估
本研究从 DNA 与 RNA 测序数据比对、转录本单基因序列比对及 BUSCO 评估三个维度,完成望春玉兰基因组组装完整性评价。 首先运用 BWA-MEM 0.7.10 软件,将 10X Genomics 与 Hi-C 双端测序数据比对至最终组装基因组;同时采用 TopHat 2.1.0 软件,把 4 种不同组织的转录组测序数据回贴至基因组序列。其次利用 Bridger 软件,设置参数--kmer length 25 --min kmer coverage 2组装得到望春玉兰转录本单基因序列,再通过 BLAT 同源比对工具将其比对至基因组支架序列。最后借助 BUSCO 软件对染色体水平基因组开展评估,系统验证参考基因组的完整度。
重复序列注释
采用同源比对结合从头预测 的策略鉴定基因组中转座子元件。同源注释层面,分别在 DNA 序列与蛋白序列水平,使用 RepeatMasker 4.0.5 与 RepeatProteinMask 软件,将基因组序列比对 Repbase 21.01 已知重复序列数据库,完成同源转座子识别。从头预测层面,依托 RepeatModeler 2.0 与 LTR Finder 1.0.6 软件构建物种特异性重复序列文库,再通过 RepeatMasker 完成全基因组转座子注释;串联重复序列则使用 TRF 4.04 软件进行预测标注。
基因结构预测
整合从头预测、同源蛋白比对、转录组证据 三类数据,借助 MAKER-P 2.31 流程完成蛋白编码基因预测。 从头预测部分:先使用 GeneMark-ES 4.32 默认参数进行自训练建模;利用 Trinity 软件以基因组引导模式拼接可变剪接转录本,设置参数--full_cleanup --jaccard_clip --no_version_check --genome_guided_max_intron 100000 --min_contig_length 200,再通过 PASA 软件校正基因结构,筛选完整基因模型用于训练 Augustus 与 SNAP 预测模型,最终在屏蔽重复序列的基因组上完成基因预测。 同源证据部分:选用拟南芥、水稻、无油樟以及近缘物种樟、鹅掌楸的蛋白序列作为比对数据集。 转录组证据部分:采用纯从头组装策略,使用 Trinity 2.0.6 组装干净转录组 reads 获得转录片段,作为表达序列标签导入 MAKER-P 流程。 经过两轮 MAKER-P 迭代整合,得到非冗余一致性基因集。 非编码 RNA 注释:利用 tRNAscan-SE 1.3.1 鉴定转运 RNA;通过 BLAST 比对 Rfam 数据库筛选小核 RNA 与微小 RNA;运用 BLASTN 比对植物已知核糖体 RNA 序列鉴定 rRNA。 最后使用 Circos 软件,在染色体上可视化标注基因密度、GC 含量、Gypsy 与 Copia 类转座子分布特征。
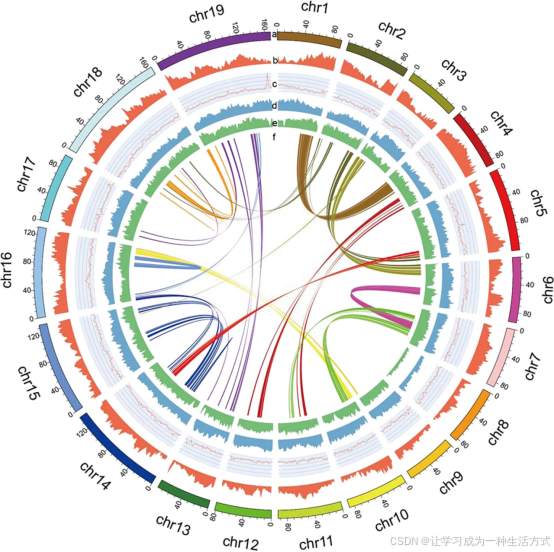
由外至内依次为:a 组装获得的拟染色体、b 基因密度、c GC 含量、d Gypsy 类转座子密度、e Copia 类转座子密度、f 染色体共线性关系
蛋白编码基因功能注释
基于序列同源性与结构域保守性,将望春玉兰预测得到的氨基酸序列比对公共数据库,完成蛋白编码基因的功能注释。 运用 BLASTP 程序(阈值e-value 设为 1e-5),将基因蛋白序列比对 KEGG、NCBI 非冗余蛋白库 NR、直系同源蛋白簇数据库 COG、SwissProt 及 TrEMBL 等主流蛋白数据库,获取最优同源匹配结果。 再借助 InterProScan 5.0 软件,整合 Pfam、SMART、PANTHER、PRINTS、ProDom 等数据库信息,完成蛋白结构域与保守基序鉴定。
基因家族鉴定
收集望春玉兰及其余 5 种被子植物(无油樟、拟南芥、樟、鹅掌楸、葡萄)的蛋白序列与核酸序列,采用 OrthoFinder 软件,以全序列两两 BLASTP 比对(e-value=1e-5)为基础构建基因家族。 通过基因序列比对 KEGG 数据库解析基因参与的代谢通路,同时结合 InterProScan 与 Pfam 注释结果,完成基因本体 GO 功能条目分类注释。
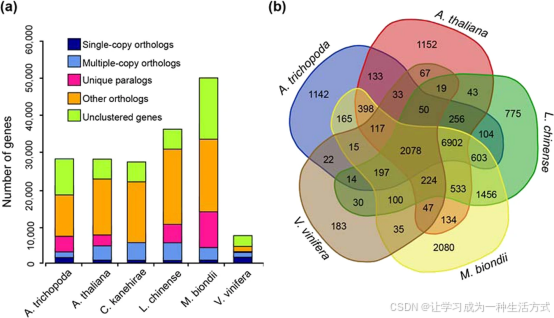
a 不同植物物种基因数量统计,结果显示望春玉兰基因数目显著高于模式植物拟南芥,同时也多于无油樟、樟、鹅掌楸、葡萄等物种。 b 韦恩图,展示望春玉兰、鹅掌楸、无油樟、拟南芥与葡萄之间共有及特有基因家族分布情况。
系统发育基因组学构建与基因家族演化分析
为明确望春玉兰与其他植物物种的基因家族亲缘关系,以及厘清木兰类植物在被子植物中的系统发育地位,本研究选取 109 个单拷贝核基因,串联其氨基酸序列构建系统发育数据集,完成 20 种种子植物的系统发育关系比对分析。
利用 OrthoFinder 软件,从 18 种被子植物(包含 2 种双子叶植物、2 种单子叶植物、2 种金粟兰科植物、8 种木兰类植物、2 种八角属植物、无油樟及睡莲)与外类群裸子植物挪威云杉中筛选直系同源基因,并与望春玉兰基因组注释蛋白编码基因进行比对。
筛选获得严格一对一直系同源基因集,使用 MAFFT 5.0 完成序列比对,再通过 GBlocks 0.91b 进行序列保守区域修剪,最后借助 Geneious 10.0.2 软件完成序列串联拼接。
筛选出物种覆盖率高于 85% 的 109 个单拷贝核基因串联氨基酸序列数据集,运用 PartitionFinder 软件以基因为单位进行数据分区筛选与最优进化模型筛选,最终得到 18 个最优分区组合。
基于该数据集,采用 RAxML-VI-HPC v.2.2.0 软件通过最大似然法 构建系统发育树,并设置 PROTGAMMAAUTO 模型开展 500 次自举重复检验,评估进化树分支置信度。
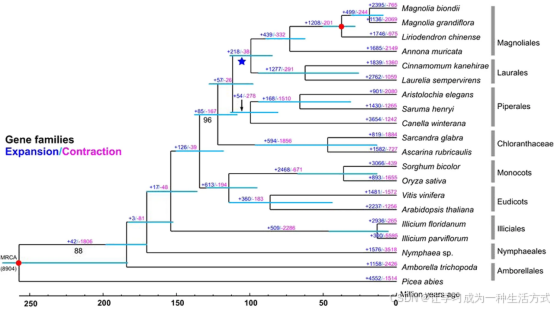
各分支节点处青绿色横条代表物种分化时间的置信区间,红色圆点表示时间校正节点,蓝色五角星标注木兰目与樟目物种共同经历的全基因组复制事件。除分支下方另行标注外,其余所有分支在最大似然法分析中均获得最高支持度。
以最优最大似然进化树为基准,借助 PAML 4 软件内置的 MCMC Tree 程序估算物种分化时间。依据 TimeTree 在线数据库设置两个分化时间校正点:鹅掌楸属与木兰属物种分化时间(3400 万 ---7700 万年前)、被子植物与裸子植物分化时间(1.68 亿 ---1.94 亿年前)。 将 OrthoFinder 聚类得到的同源基因簇与 RAxML 构建的系统发育树拓扑结构导入 CAFE 4.2 软件,分析各物种不同基因家族发生显著扩张与收缩的演化特征。
基因组共线性与全基因组复制事件分析
望春玉兰基因组共注释得到 47547 个蛋白编码基因,为探究其基因数量繁多的演化成因,本研究开展全基因组复制事件分析。 葡萄基因组已证实发生过一次全基因组三倍化事件,同科的鹅掌楸也已报道存在一次全基因组复制事件。本研究提取望春玉兰、鹅掌楸与葡萄的蛋白编码序列及对应 CDS 序列,使用 Python 版 MCscanX 软件开展基因组共线性分析,设定共线性区块至少包含 5 个同源基因对。 通过绘制共线性点阵图,统计基因组各区域共线性深度,比对判定望春玉兰与其他被子植物的古倍性水平。 进行同义替换率 Ks 分布分析时,整理 OrthoMCL 基因家族聚类结果,筛选出两物种均仅含单基因成员、以及任一物种含双基因成员的基因家族,再利用 PAML 软件计算同源基因对的同义替换率 Ks 值。
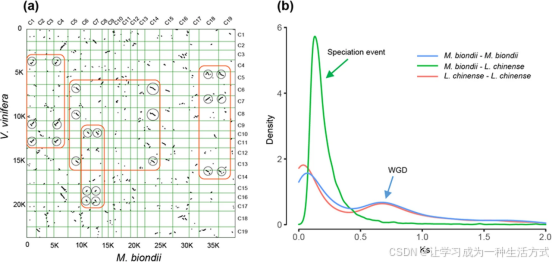
a 望春玉兰与葡萄基因组共线性比对:同源基因点阵图显示,望春玉兰基因组与葡萄基因组整体呈现2 对 3 的染色体对应关系。 b 基于 OrthoMCL 基因家族聚类结果,统计望春玉兰与鹅掌楸物种内旁系同源基因、物种间直系同源基因的同义替换率(Ks)分布特征。
萜类合成酶基因鉴定与表达分析
本研究选取无油樟、拟南芥两个物种,与望春玉兰开展萜类合成酶(TPS)基因家族比较基因组分析,两物种已注释 TPS 基因序列均调取自 Chaw 等人发表的数据集。
以 Pfam 数据库中PF03936、PF01397 两大保守结构域为检索序列,利用 HMMER 3.0 软件(阈值e-value=1e-5)在望春玉兰蛋白组序列中筛选候选 TPS 基因,最终鉴定得到 102 条 TPS 候选蛋白序列。
使用 MAFFT 5.0 完成多序列比对,借助 MEGA 4.0 手动优化比对结果,采用 IQ-TREE 软件构建系统发育树,并设置 1000 次自举检验评估分支可信度。
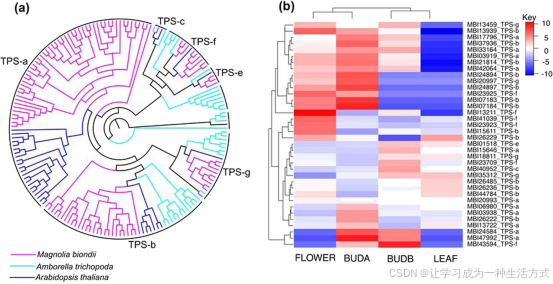
a 无油樟(38 个)、拟南芥(32 个)与望春玉兰(102 个)萜类合成酶(TPS)基因系统发育树。 b 基于幼叶、盛花、减数分裂前花蕾、减数分裂后花蕾四组转录组数据绘制的 TPS 基因差异表达热图。
利用 Bowtie2 软件将转录组测序 reads 比对至基因集蛋白编码序列,借助 eXpress 软件统计各组织基因表达量,再通过 edgeR 软件筛选差异表达基因。 设定筛选阈值:错误发现率 FDR<0.001,对数倍变化值\(\log_2\text{FC}>2\)或\(\log_2\text{FC}<-2\),以此筛选望春玉兰中差异表达的 TPS 基因。
数据公开
本研究基因组组装序列、注释文件及其他辅助数据已上传至 Dryad 数据库,访问链接:https://doi.org/10.5061/dryad.s4mw6m947;原始测序数据存入国家基因库,入库编号:CNP0000884。
研究结果
测序数据概况
本研究共获得 33 倍 PacBio 单分子长读长数据,总数据量 66.78 Gb,平均读长 10.32 kb;80 倍 10X Genomics 双端短读长数据,总量 175.45 Gb;Hi-C 测序数据约 153.78 Gb。 转录组测序分别对幼叶、盛花、减数分裂前后两个时期花蕾测序,原始数据量依次为 4.62 Gb、4.60 Gb、4.67 Gb、4.73 Gb。
基因组大小与杂合度评估
k-mer 频数统计结果显示,主峰深度为 48,预估基因组大小 2.17 Gb; 利用 GCE 软件分析得出 k-mer 主峰深度 29,推算基因组大小 2.24 Gb,基因组杂合度 0.73%,重复序列占比 61.83%。 望春玉兰是目前已完成测序的木兰类植物中基因组规模最大的物种。
基因组组装与质量评价
采用 Miniasm 软件完成初步组装,得到包含 15713 条 Contig、总长 2.20 Gb 的基因组草图,Contig N50 为 267.11 kb。 先后经过三轮 PacBio 长序列纠错、一轮 10X Genomics 数据校正,最终得到 Contig 版本基因组:序列总长 2.22 Gb,共 15615 条 Contig,Contig N50 达 269.11 kb。 借助 Hi-C 数据完成染色体挂载,总计 89.17% 的序列成功锚定至 19 条染色体上,挂载序列总长 1.98 Gb,基因组内未知碱基 N 总长 7365981 bp,占基因组总长 0.33%;剩余 9455 条 Contig(0.24 Gb)未能完成定位。 再结合 PacBio 与 10X 数据进一步优化支架序列,最终获得 9510 条 Scaffold,组装基因组总长 2.23 Gb,Scaffold N50 为 92.86 Mb,组装结果与 k-mer 预估基因组大小高度吻合。
组装质量多维度验证结果:
- 二代测序数据比对:10X Genomics、Hi-C 数据比对率分别为 98.40%、92.50%;测序深度统计显示,基因组中超过 98.04% 的碱基 10X 数据覆盖度大于 10 倍,86.00% 碱基 Hi-C 数据覆盖度大于 10 倍。
- 转录组数据回贴:叶片、盛花、前后两个时期花蕾的转录组序列比对率分别为 93.3%、94.4%、92.9%、93.7%。
- 转录本序列比对:组装基因组可覆盖 86.88% 的全长转录本序列。
- BUSCO 完整性评估:在 1375 条陆生植物保守单拷贝基因中,完整基因占比 88.50%(完整单拷贝基因 85.20%、完整多拷贝基因 3.30%),片段化基因占比 4.40%。 以上结果证明望春玉兰参考基因组组装完整度良好。
重复序列注释
望春玉兰基因组中重复序列总长 1478819185 bp,占基因组全长 66.48%,其中长末端重复序列(LTR)为最主要重复元件,占基因组总长 58.06%。 两大主流 LTR 超家族序列特征:Copia 类总长 659463750 bp,占 LTR 序列总长 45.26%;Gypsy 类总长 727531048 bp,占比 50.66%。 分布规律:Gypsy 转座子密度与基因密度呈负相关,Copia 转座子在基因组中均匀分布,与基因分布无明显关联。 其余重复元件占比:DNA 转座子 5.86%、卫星序列 0.24%、简单重复序列 0.79%、其他类型重复序列 0.32%。
基因预测与功能注释
共注释获得 47547 个蛋白编码基因,同时鉴定出 109 个微小 RNA、904 个转运 RNA、1918 个核糖体 RNA、7426 个小核 RNA。 望春玉兰蛋白编码基因平均全长 10980 bp,平均编码区序列长度 957 bp,单基因平均外显子数量 4.4 个。 与无油樟、拟南芥、樟、鹅掌楸、水稻五种被子植物对比发现,望春玉兰基因总数最多,平均内含子长度约 2774 bp 亦为最高,与自身偏大的基因组特征相符;但其基因全长、内含子长度中位数偏低,说明存在大量超长内含子基因拉高了整体平均值。
功能注释结果显示,47547 个蛋白编码基因中共有 39111 个基因获得明确功能注释,注释率达 82.26%;剩余约 17.74% 未注释基因,一部分源于组装与注释误差,另一部分大概率为物种特有新功能基因。
基因家族分析
望春玉兰基因组共鉴定得到 15089 个基因家族,其中物种特有基因家族 1928 个,包含特有基因 10280 个。 五物种共有基因家族 2078 个,涵盖望春玉兰、鹅掌楸、无油樟、拟南芥、葡萄。
对望春玉兰特有基因家族进行 KEGG 富集分析,结果显著富集于核苷酸代谢、植物 - 病原菌互作、生物碱、泛醌、萜醌类及苯丙烷类等次生代谢物质合成通路,与该物种富含萜类、酚类、生物碱类活性物质的生物学特征高度契合。 GO 功能富集显示,特有基因主要富集于分子结合功能、核酸结合、环状化合物结合、水解酶活性等功能条目。 这些参与次生代谢合成与植物抗病互作的特有基因,能够增强植株抵御病原菌能力,同时助力植株与其他生物建立有益共生关系。
系统发育基因组学分析
基于 109 个单拷贝核基因,以 19 种被子植物 + 裸子植物挪威云杉为外类群构建进化树,拓扑结构稳定可靠。 进化关系明确:木兰类植物与金粟兰科植物互为姊妹群 (后验概率 96%),二者共同构成单子叶植物与双子叶植物演化支的外类群(后验概率 100%)。 物种分化时间估算:木兰目与樟目亲缘关系最近,分化时间约 9900 万年前(8400 万 ---1.16 亿年前);木兰目内部木兰科与番荔枝科分化于约 7300 万年前;鹅掌楸属与木兰属分化时间约 3800 万年前。
基因家族演化
利用 CAFE 软件分析基因家族扩张与收缩事件,望春玉兰基因组中 15683 个基因家族里,2395 个基因家族发生显著扩张,765 个基因家族发生显著收缩。 扩张基因家族 KEGG 富集结果集中在代谢通路、次生代谢物质合成、植物激素信号转导、ABC 转运蛋白通路等; GO 功能富集主要涉及离子结合、转移酶活性、物质代谢、细胞进程、氧化还原酶活性、应激响应等功能。 此类基因尤其是次生代谢合成、激素调控、逆境响应相关基因的大量扩张,提升了望春玉兰的环境适应能力与生态竞争优势。
基因组共线性与全基因组复制事件分析
望春玉兰基因组内部筛选得到 147 个共线性区块,包含 1738 对同源基因; 与鹅掌楸比对得到 393 个共线性区块、13674 对同源基因,二者长片段共线性区域基本呈现 1:1 对应关系,证实二者演化历程高度相近,推测木兰属与鹅掌楸属在被子植物共同祖先分化后,共同经历过一次全基因组复制事件; 望春玉兰与葡萄基因组共线性片段呈现稳定 2:3 对应模式,进一步佐证木兰类物种演化历程中发生过独立全基因组复制事件。
同源基因同义替换率 Ks 分布显示,望春玉兰物种内旁系同源基因 Ks 主峰约为 0.70,对应分化年代约 1.16 亿年前,与鹅掌楸 Ks 峰值高度重合。 说明二者要么源自共同祖先的同一次全基因组复制事件,要么在相近演化时期各自发生独立复制事件; 而两属物种直系同源基因 Ks 峰值约 0.15,分化时间约 2480 万年前,晚于全基因组复制发生时间,证明此次全基因组复制事件至少为木兰科两大属物种所共有。
萜类合成酶基因
望春玉兰花蕾中提取的挥发油主要成分为萜类化合物,这类物质均由萜类合成酶(TPS)催化合成。本研究在望春玉兰基因组中共鉴定出102 个 TPS 候选基因,数量与樟的 101 个 TPS 基因相近。
为明确望春玉兰 TPS 家族分类,整合望春玉兰、拟南芥、无油樟 TPS 蛋白序列构建系统发育树,可将其划分为 6 个亚家族:TPS-a(52 个)、TPS-b(27 个)、TPS-c(1 个)、TPS-e(3 个)、TPS-g(10 个)、TPS-f(9 个) 。
结合幼叶与三个不同发育时期花组织的转录组表达谱分析,共筛选出 36 个高表达 TPS 基因,各亚家族数量依次为 TPS-a 11 个、TPS-b 13 个、TPS-c 1 个、TPS-e 1 个、TPS-f 6 个、TPS-g 4 个。其中 33 个基因(TPS-a、TPS-b 各 10 个)在花组织中的表达量显著高于叶片,表明这类基因大概率参与望春玉兰花朵生长发育过程中各类萜类物质的代谢合成。
讨论
k-mer 分析结果显示望春玉兰基因组规模偏大、结构复杂,预估基因组大小 2.24 Gb,杂合度 0.73%,重复序列占比 61.83%。相较于杂合度仅 0.087% 的丛生画眉草,其基因组杂合度高出近十倍,也是造成基因组组装连续性偏低的重要原因。
本研究仅采用 33 倍 PacBio 长读长数据完成初步组装,最终得到总长 2.23 Gb、含 15615 条 Contig 的基因组草图,Contig N50 仅 269.11 kb。较短的 Contig 片段预示组装序列碎片化程度较高,进而影响后续 Hi-C 染色体挂载的组装质量。 利用 Hi-C 数据进行染色体锚定过程中,总计 6899 条总长 1.00 Gb 的 Contig 发生序列断裂,最终舍弃 0.18 Gb 序列;经 Juicebox 软件手动校正 Hi-C 互作图谱后,仍有 6911 条 Contig 结构受损、2358 个基因结构被打乱,0.24 Gb 序列无法完成染色体定位。 BUSCO 评估结果也显示,相较于 Contig 水平基因组,Scaffold 版本完整保守基因占比下降、片段化基因占比上升。 因此本研究后续仅将 Hi-C 挂载基因组用于染色体共线性分析,其余各类比较基因组分析均采用 Contig 组装版本开展。 同时,望春玉兰注释得到数量庞大的蛋白编码基因,一方面源于基因组组装碎片化导致的基因拆分注释,另一方面也与基因组内高含量转座子密切相关;相较于同科鹅掌楸,其编码序列平均长度与中位长度显著偏短,也印证了这一结论。
本研究获得的望春玉兰染色体水平参考基因组,明确了 19 条染色体上的基因分布、重复元件组成与基因组结构特征,不仅为该物种分子基础研究与实际育种应用提供核心遗传资源,也为木兰属及近缘物种演化、比较基因组学研究奠定基础。
基于涵盖木兰类四大目中三类代表物种的 20 种种子植物、109 个单拷贝核基因开展系统发育基因组学分析,结果高度支持木兰类植物与金粟兰科互为姊妹群 ,二者共同构成单子叶植物与双子叶植物演化支的外类群。 该拓扑结构与叶绿体基因组系统发育结果、多基因联合建树结论一致,但与仅以樟为木兰类唯一代表的被子植物系统发育研究、92 种链形植物及 20 种被子植物转录组系统发育研究结论不同(后两者认为木兰类与双子叶植物为姊妹群)。 本研究建树结果可靠性更高,主要原因有三点:
- 选用保守性更强、更少发生趋同演化的氨基酸序列,避开易出现替换饱和的密码子第三位核苷酸序列;
- 针对大规模串联序列数据集,采用最优数据分区策略并精准筛选进化模型;
- 物种取样覆盖面广,囊括被子植物八大主要演化支系(仅缺少无基因组数据的金鱼藻目)。 后续研究进一步完善物种取样、扩充基因数据集并优化分析方法,将能更精准厘清基部被子植物的分化次序。
本次组装结果进一步完善了学界对木兰类植物全基因组复制事件发生时间的认知。基因组共线性分析与同义替换率 Ks 分布分析证实木兰目物种曾发生一次全基因组复制事件,该事件发生时间约 1.16 亿年前,与前人研究结果相符。 结合物种分化时间来看,木兰目与樟目分化时间约 8400 万 ---1.16 亿年前,分化均值约 9900 万年前,与全基因组复制发生时间高度接近;结合木姜子属物种基因组演化研究推测,此次复制事件大概率发生在两目物种分化前夕,但该推论仍存在争议,例如同为木兰目的刺果番荔枝并未检测到近期全基因组复制事件,仍需更多物种数据佐证。
望春玉兰作为药用植物,花蕾中富含倍半萜、单萜类物质组成的药用挥发油,而在中部被子植物中,TPS-a 亚家族主要负责合成倍半萜,TPS-b 亚家族主导单萜合成 。 通过对比多种被子植物 TPS 基因家族组成与进化树结构,证实望春玉兰 TPS 家族发生明显扩张,尤以 TPS-a、TPS-b 亚家族最为显著。 组织表达模式显示,大量 TPS-a、TPS-b 家族基因在花组织中特异性高表达,与该物种花蕾挥发油中高含量倍半萜、单萜类活性成分的积累特征完全吻合。
结论
本研究整合 10X Genomics、PacBio 长读长测序与 Hi-C 染色体挂载技术,成功组装获得望春玉兰参考基因组。该基因组大小约 2.22 Gb,基因组杂合度 0.73%,重复序列占比 66.48%,是目前已测序的六种木兰类植物中基因组规模最大的物种。 研究共注释得到 47547 个蛋白编码基因,基因功能注释完成率达 82.26%。 系统发育分析明确证实木兰类植物与金粟兰科互为姊妹群,二者共同作为单双子叶植物演化支的外类群。 该基因组数据不仅有助于深入解析木兰属植物遗传多样性、特有性状形成的分子机制,也为被子植物早期辐射演化机制研究与药用木兰分子育种工作提供重要理论依据与数据支撑。