目录
单细胞的数据格式学习:单细胞 10X 和seurat对象学习-CSDN博客
①数据读取
数据集:GSE164241

rm(list = ls())
library(Seurat)
folders=list.files('./',pattern='[123]$')
folders
scList = lapply(folders,function(folder){
CreateSeuratObject(counts = Read10X(folder),
project = folder,
min.cells = 3, min.features = 200)
})
BM <- merge(scList[[1]],
y = c(scList[[2]],scList[[3]]),
add.cell.ids = c("BM1","BM2","BM3"),
project = "BM")
BM
GM <- merge(scList[[4]],
y = c(scList[[5]],scList[[6]]),
add.cell.ids = c("GM1","GM2","GM3"),
project = "GM")
GM##两部分数据进行读取合并②计算线粒体基因比例
-
细胞基因检出数,低质量细胞基因检出数通常较低,双细胞或者同时捕获多个细胞会有很高的基因数。所以要去除低质量的,和过高的细胞。
-
细胞检测出的分子数
-
线粒体基因比例,一般低质量细胞或者死细胞线粒体基因检出数很高。但是特殊情况特殊对待,有些细胞功能活跃,线粒体活跃,检出数自然也会很高。所以不能一刀切。
GM[["percent.mt"]] <- PercentageFeatureSet(GM,pattern = "^MT-")#分别计算线粒体
BM[["percent.mt"]] <- PercentageFeatureSet(BM,pattern = "^MT-")
preQC_GM <- VlnPlot(GM, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3,
group.by = "orig.ident",
pt.size = 0)
preQC_BM <- VlnPlot(BM, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3,
group.by = "orig.ident",
pt.size = 0)
preQC_GM
preQC_BM
dev.off()
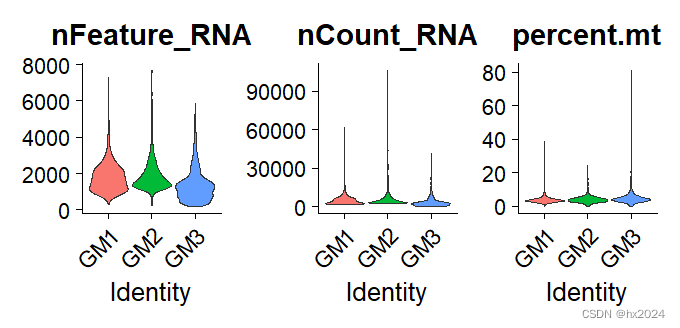
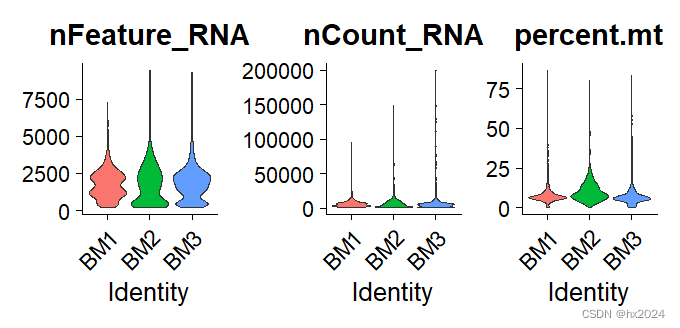
③分开进行质控
nCount_RNA:每个细胞的UMI数量
nFeature_RNA:基因数
percent.mt:线粒体基因百分比
GM、BM分开,涉及后期QC参数有可能不同。
单细胞质控是指对单细胞测序数据进行质量控制和过滤,保证数据的准确性和可靠性。单细胞测序数据存在许多干扰因素,如PCR扩增偏差、杂交、RNA降解等,这些因素可能导致数据出现噪声、低表达、脱落细胞等问题。因此,单细胞质控的主要任务是过滤出质量高、可靠的单细胞数据,以便进行下一步的数据分析和挖掘。
单细胞质控通常包括以下几个方面的内容:
-
基础质控:包括测序质量、mapping率、reads数、UMI数等基本指标的计算和分析。
-
细胞质控:包括细胞表达量、基因检测率、rRNA比例、mapping率等细胞特异性指标的计算和分析,用于确定是否保留该细胞的数据。
-
基因质控:包括基因表达量、基因检测率、基因表达异质性等指标的计算和分析,用于确定保留哪些基因。
-
附加质控:包括PCA分析、t-SNE分析、聚类分析等附加分析,用于确定哪些细胞可以被合并或排除。
#设定各指标阈值使用subset函数取子集
#进行质控
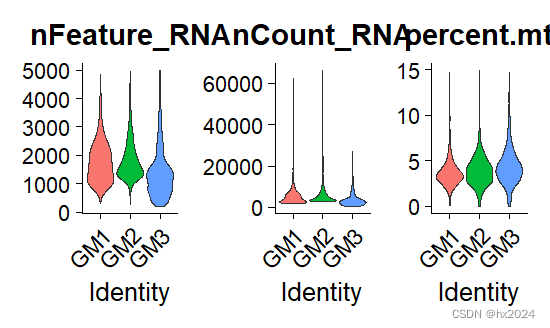
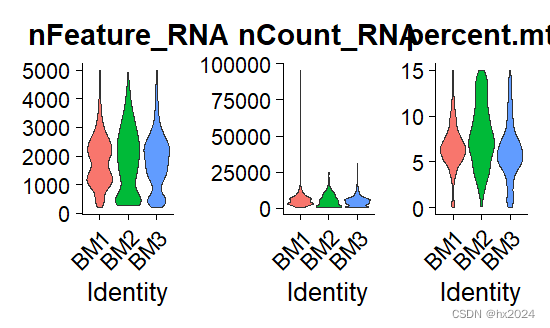
GM <- subset(GM, subset = nFeature_RNA > 200 & nFeature_RNA < 5000 & percent.mt < 15)
BM <- subset(BM, subset = nFeature_RNA > 200 & nFeature_RNA < 5000 & percent.mt < 15)
postQC_GM <- VlnPlot(GM, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3,
group.by = "orig.ident",
pt.size = 0)
postQC_BM <- VlnPlot(BM, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3,
group.by = "orig.ident",
pt.size = 0)
postQC_GM#这时线粒体基因比例都小于15%
postQC_BM
接下来就是将两个数据合并,去除批次效应,整合成一个seurat对象进行下游降维。
④两组单细胞数据合并
#FindIntegrationAnchors合并数据
BM <- NormalizeData(BM)
BM <- FindVariableFeatures(BM, nfeatures = 4000)#数据标准化及计算高变基因
GM <- NormalizeData(GM)
GM <- FindVariableFeatures(GM, nfeatures = 4000)
#整合成一个seurat对象进行下游降维,IntegrateData去除批次效应
sampleList <- list(GM, BM)
scedata <- FindIntegrationAnchors(object.list = sampleList, dims = 1:50)
scedata <- IntegrateData(anchorset = scedata, dims = 1:50)
save(scedata, file = "scedata.RData")⑤细胞周期评分
作者还计算了细胞周期评分,因为我们收集到的细胞可能处于不同的分裂时期,所以看周期是很有必要的,尤其是针对具体的研究目的。
rm(list = ls())
library(Seurat)
load("scedata.RData")
s.genes <- cc.genes$s.genes#细胞周期查看(根据需求)
g2m.genes <- cc.genes$g2m.genes
scedata <- CellCycleScoring(scedata,
s.features = s.genes,
g2m.features = g2m.genes,
set.ident = TRUE)
VlnPlot(scedata,features = c("S.Score","G2M.Score"),group.by = "orig.ident")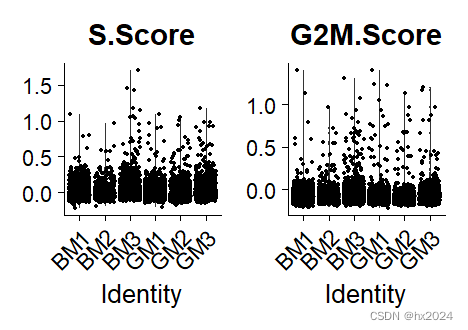
⑥降维标准流程
降维
单细胞数据RunPCA降维分析前的scale操作,通常指对数据进行标准化或归一化处理,旨在消除不同基因或细胞之间的量纲或数量级的差异。这样做可以使得不同基因或细胞在降维后对PCA的贡献更加均衡,避免了那些具有高表达量的基因或高测序深度的细胞对PCA结果产生过大影响的情况。数据标准化与wilcox分析_wilcox检验的数据必须log2吗-CSDN博客
# 标准流程(vars.to.regress进行降维)
scedata <- ScaleData(scedata, vars.to.regress = c("S.Score", "G2M.Score"), verbose = FALSE)
scedata <- RunPCA(scedata, npcs = 50, verbose = FALSE)
scedata <- FindNeighbors(scedata, reduction = "pca", dims = 1:50)
scedata <- FindClusters(scedata,
resolution = seq(from = 0.1,
to = 1.0,
by = 0.1))
scedata <- RunUMAP(scedata, reduction = "pca", dims = 1:50)
library(clustree)
clustree(scedata)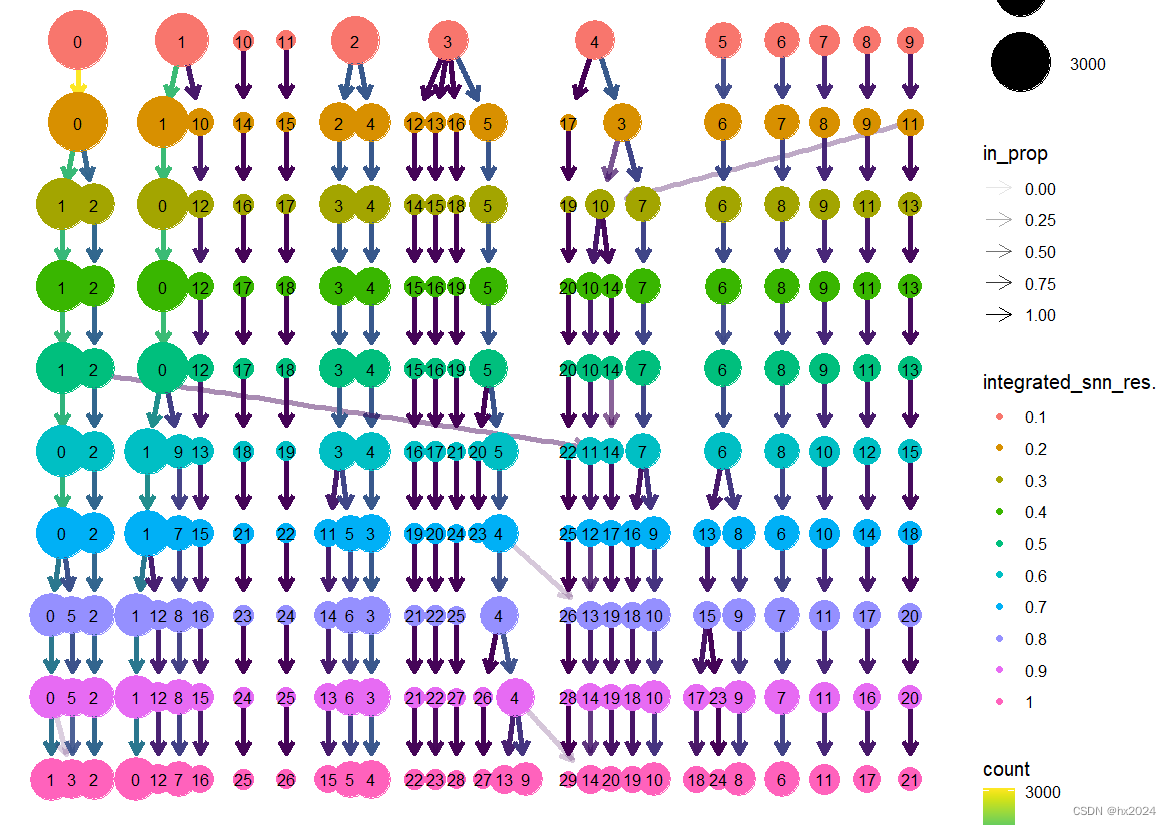
PCA流程结束。我们需要挑选合适的PCs进行后续的细胞亚群鉴定
这里官方给出了两种方法,JackStraw()和ElbowPlot()
实际上该选择多少并没有一个明确的规定,往往只能通过继续向下游分群注释去做,出现问题了回来调整或者往下做之后更换几个PC再做一次看结果的重复性是否良好
scedata <- JackStraw(scedata, num.replicate = 100)#JackStraw方法
scedata <- ScoreJackStraw(scedata, dims = 1:20)
JackStrawPlot(scedata, dims = 1:15) #可视化前15个PC
ElbowPlot(scedata)#肘型图
##默认都是计算50个PC可根据需求调整参数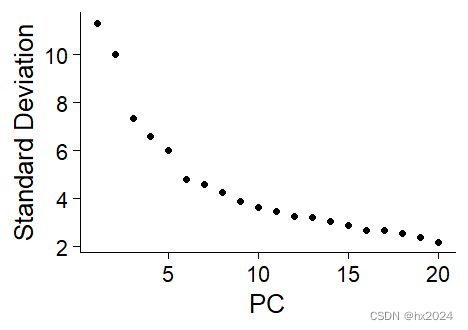
关于单细胞降维最佳PC数量选取 - 简书 (jianshu.com)的筛选。
1.主成分累积贡献大于90%;2.PC本身对方差贡献小于5%;3.两个连续PCs之间差异小于0.1%
UMAP可视化
DimPlot(scedata)#UMAP降维进行可视化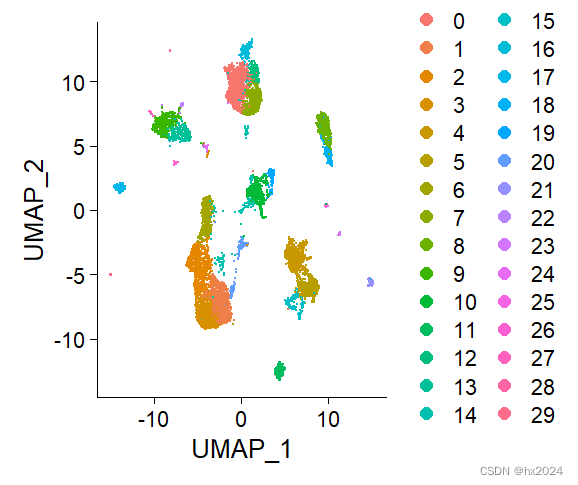
选择分群
#选择好合适的细胞分群,将其设置为active.ident
Idents(scedata) <- "integrated_snn_res.1"#这里根据上面的分析进行选择
scedata$seurat_clusters <- scedata@active.ident
DimPlot(scedata,label = T,
split.by = "orig.ident",ncol = 3)#每个样本进行性可视化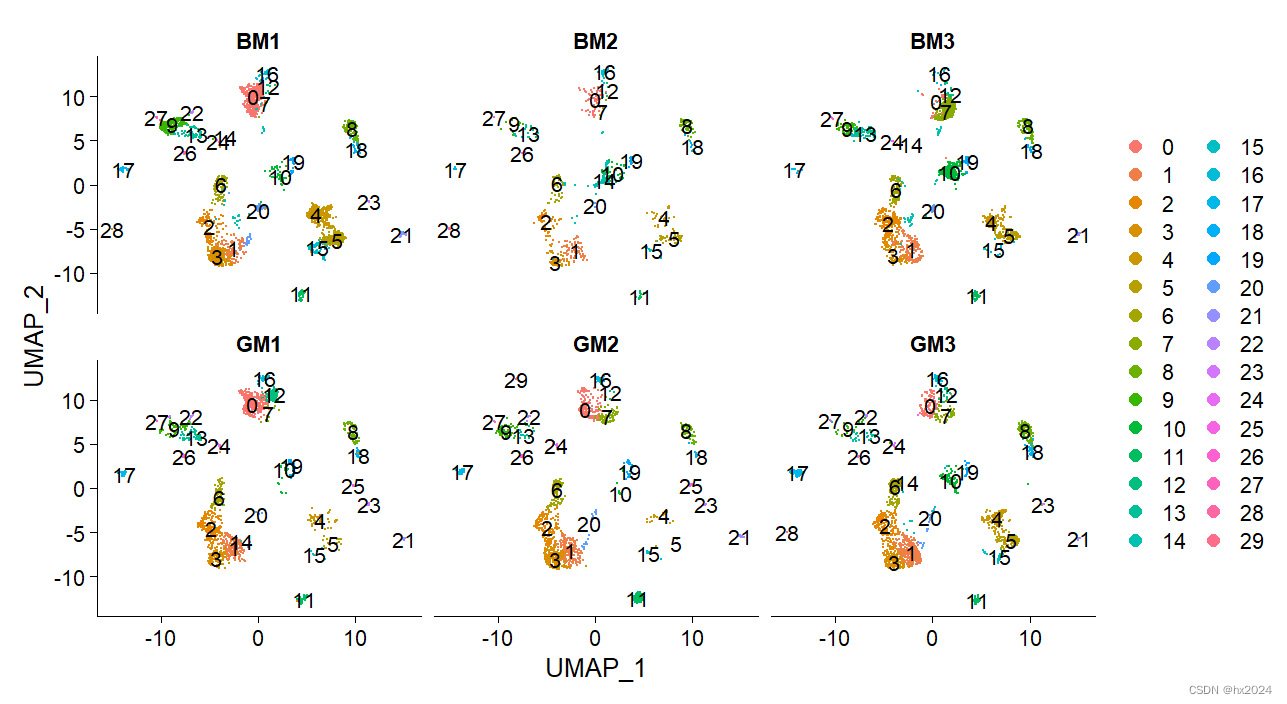
⑦marker基因
分析marker基因
#使用FindAllMarkers鉴定各个细胞群的高表达基因
DefaultAssay(scedata) <- "RNA"
all.markers <- FindAllMarkers(scedata,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.75)
significant.markers <- all.markers [all.markers $p_val_adj < 0.2, ]
#write.csv(significant.markers, file = "significant.markers.csv")#保存marker基因可视化
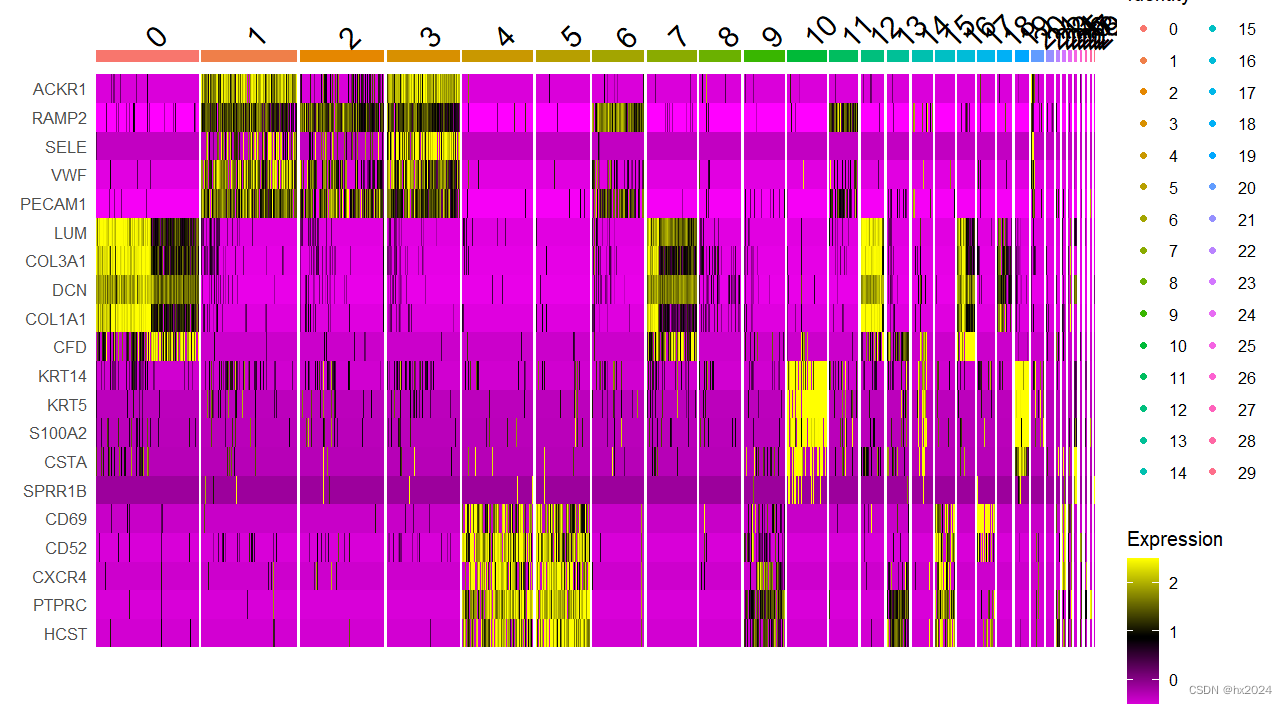
markers <- c("ACKR1","RAMP2","SELE","VWF","PECAM1",
"LUM","COL3A1","DCN","COL1A1","CFD",
"KRT14","KRT5","S100A2","CSTA","SPRR1B",
"CD69","CD52","CXCR4","PTPRC","HCST")
DotPlot(scedata,features = markers)+coord_flip()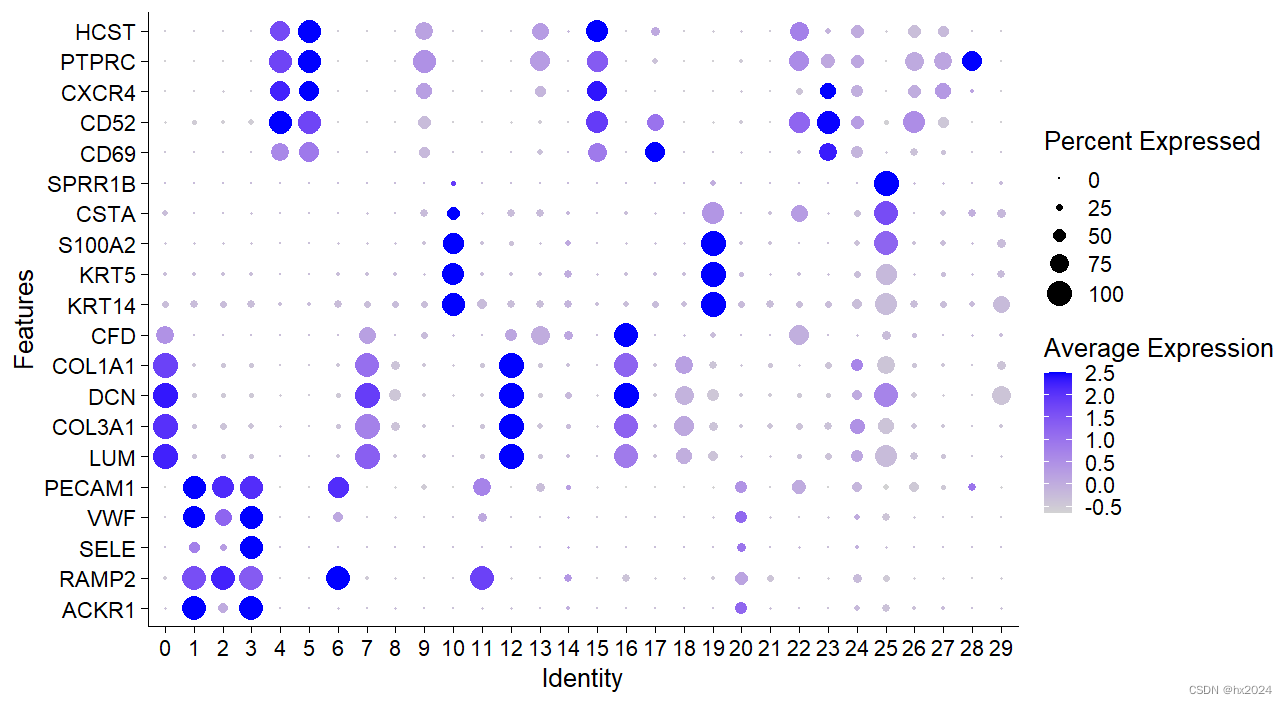
#UMAP图
FeaturePlot(scedata,features = c("ACKR1","LUM","KRT14","CD69"))
#热图
alldata <- ScaleData(scedata,
features = markers,
assay = "RNA")
p <- DoHeatmap(alldata,
features = markers,
group.by = "seurat_clusters",
assay = "RNA")
p
dev.off()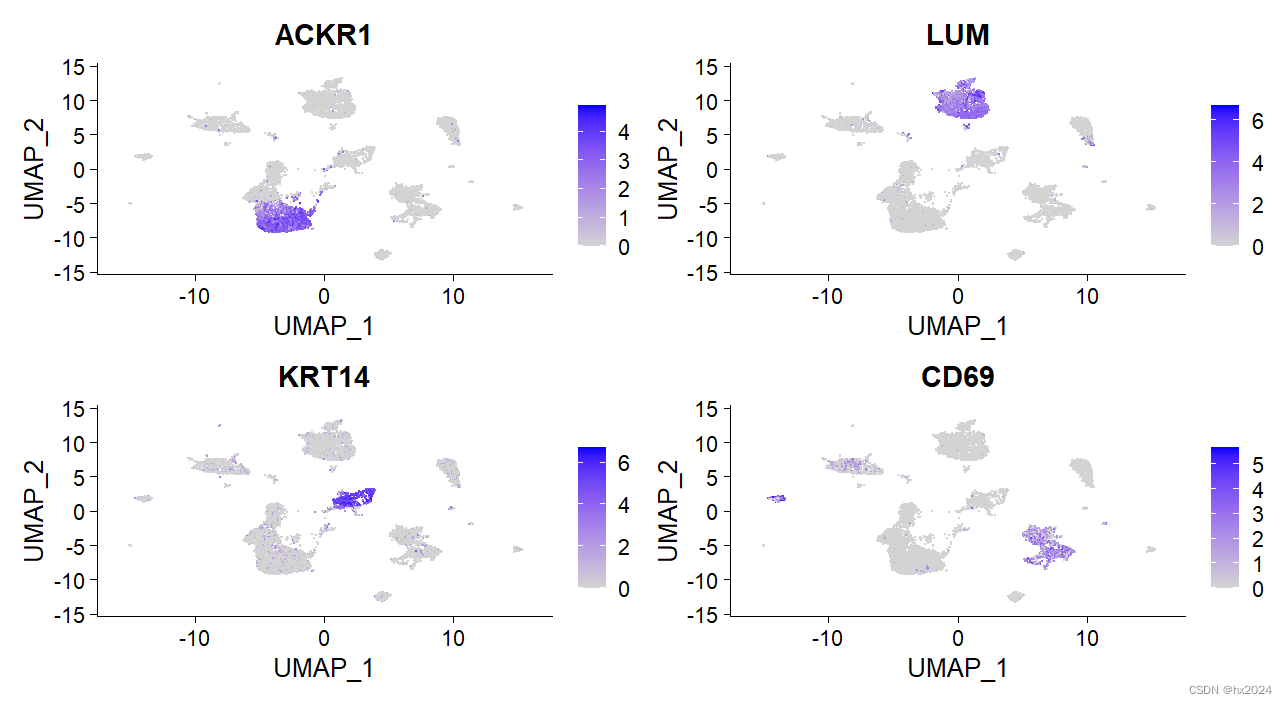
⑧细胞定群命名
#对各个细胞群命名:自动定群结果不一定完全正确
scedata <- subset(scedata, idents = c("21"), invert = TRUE)#去掉低质量细胞群
new.cluster.ids <- c("0"="Fibroblast",
"1"="Endothelial",
"2"="Endothelial",
"3"="Endothelial",
"4"="Immune",
"5"="Immune",
"6"="Endothelial",
"7"="Fibroblast",
"8"="Other",
"9"="Immune",
"10"="Epithelial",
"11"="Endothelial",
"12"="Fibroblast",
"13"="Immune",
"14"="Other",
"15"="Immune",
"16"="Fibroblast",
"17"="Endothelial",
"18"="Fibroblast",
"19"="Epithelial",
"20"="Endothelial",
"22"="Immune",
"23"="Immune",
"24"="Immune",
"25"="Epithelial",
"26"="Immune",
"27"="Immune",
"28"="Immune",
"29"="Other")
scedata <- RenameIdents(scedata, new.cluster.ids)
scedata$celltype <- scedata@active.ident
DimPlot(scedata, group.by = "celltype")
save(scedata, file = "scedata1.RData")##进行降维和清洗后的数据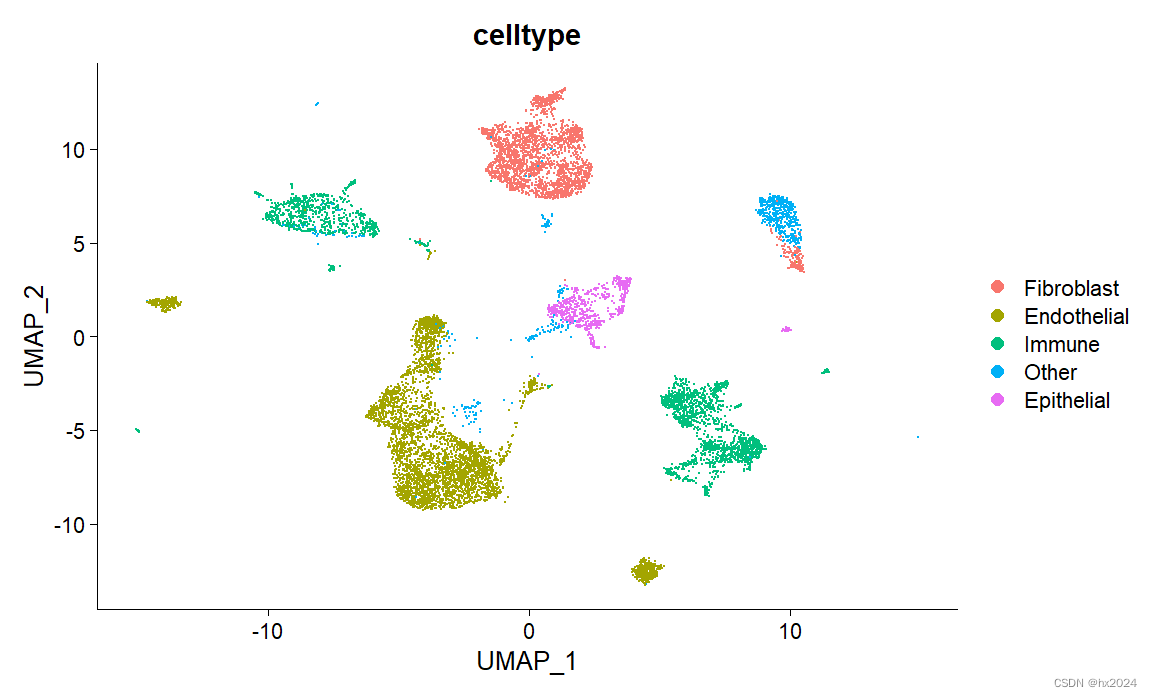
感谢:TS的美梦
跟着Cell学单细胞转录组分析(八):单细胞转录组差异基因分析及多组结果可视化_单细胞测序差异基因显著性怎么看-CSDN博客
感谢:Biomamba-生信基地的个人空间-Biomamba-生信基地个人主页-哔哩哔哩视频 (bilibili.com)