今天被问到了一个关于sc.tl.rank_genes_groups()的奇怪的问题
import scanpy as sc
import pandas as pd
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
# from CellDART import da_cellfraction
# from CellDART.utils import random_mix
from sklearn.manifold import TSNE
ref_adata = sc.read_h5ad("./scRNA.h5ad")
ref_adata
ref_adata.var_names_make_unique()
sc.pp.normalize_total(ref_adata)
sc.pp.log1p(ref_adata)
#PCA and clustering
sc.tl.pca(ref_adata, svd_solver='arpack')
sc.pp.neighbors(ref_adata, n_neighbors=6, n_pcs=40)
sc.tl.umap(ref_adata)
sc.tl.leiden(ref_adata, resolution = 0.5)
sc.pl.umap(ref_adata, color=['leiden','cellType'])
sc.tl.rank_genes_groups(ref_adata, 'cellType', method='wilcoxon')
sc.pl.rank_genes_groups(ref_adata, n_genes=15, sharey=False)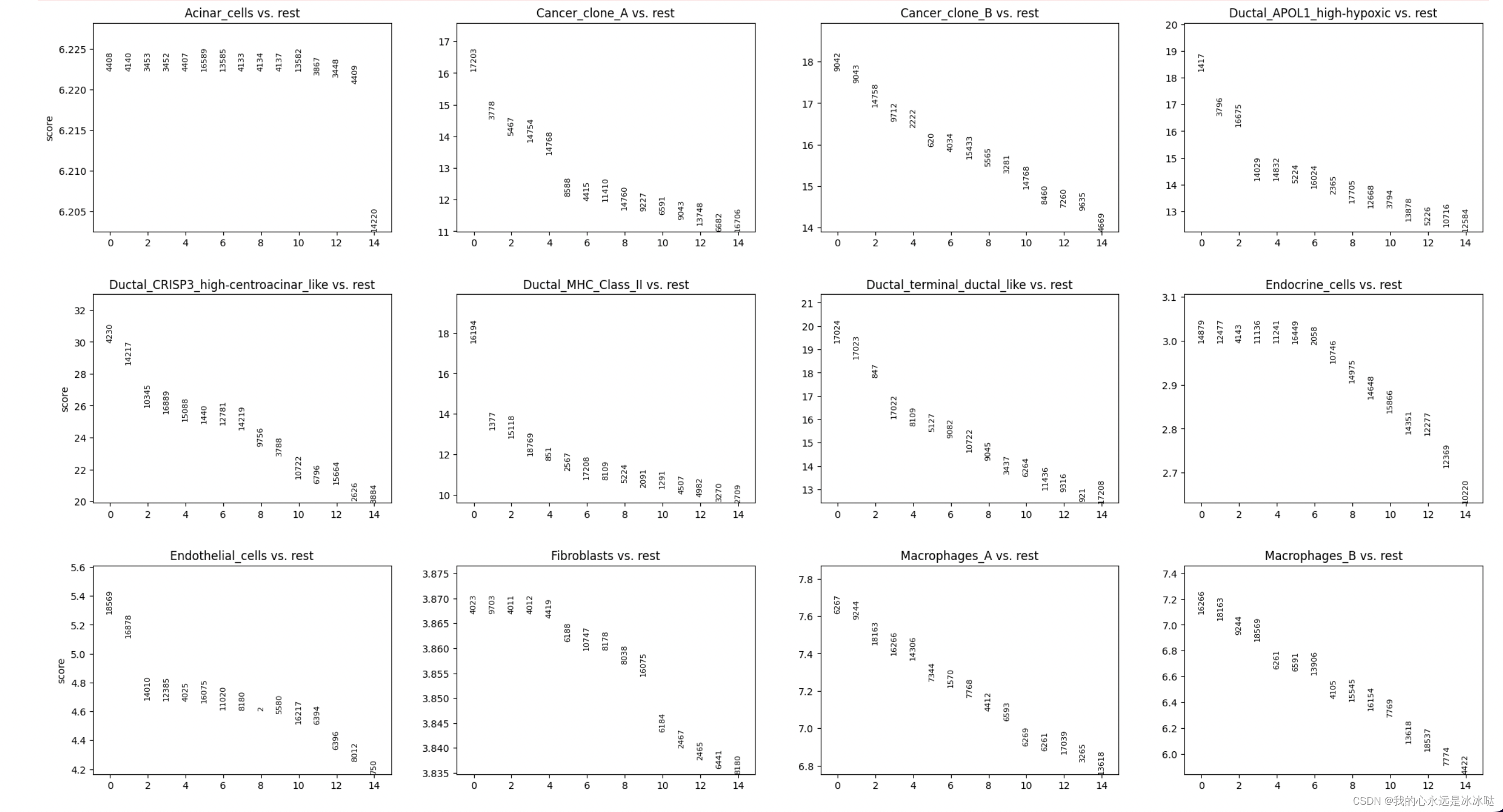
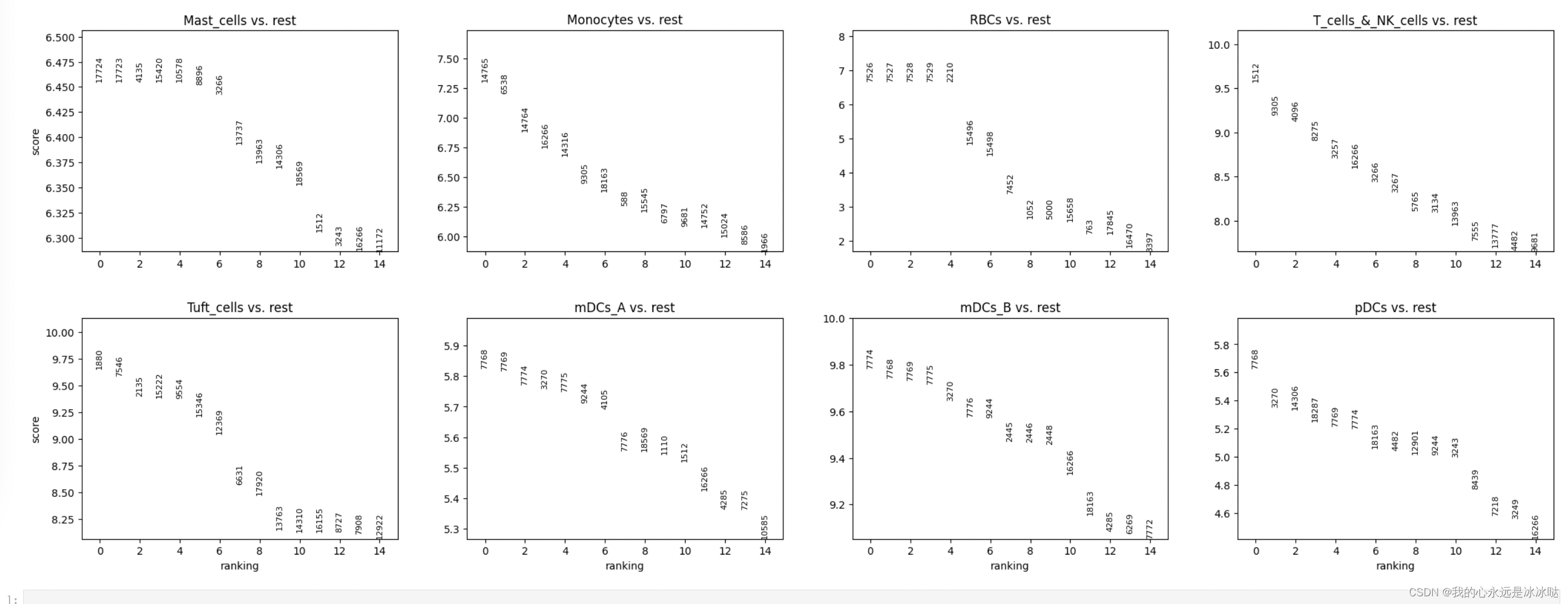
可以看到这里的检验结果是以数字的形式显示,而不是以基因的形式显示的
解决办法
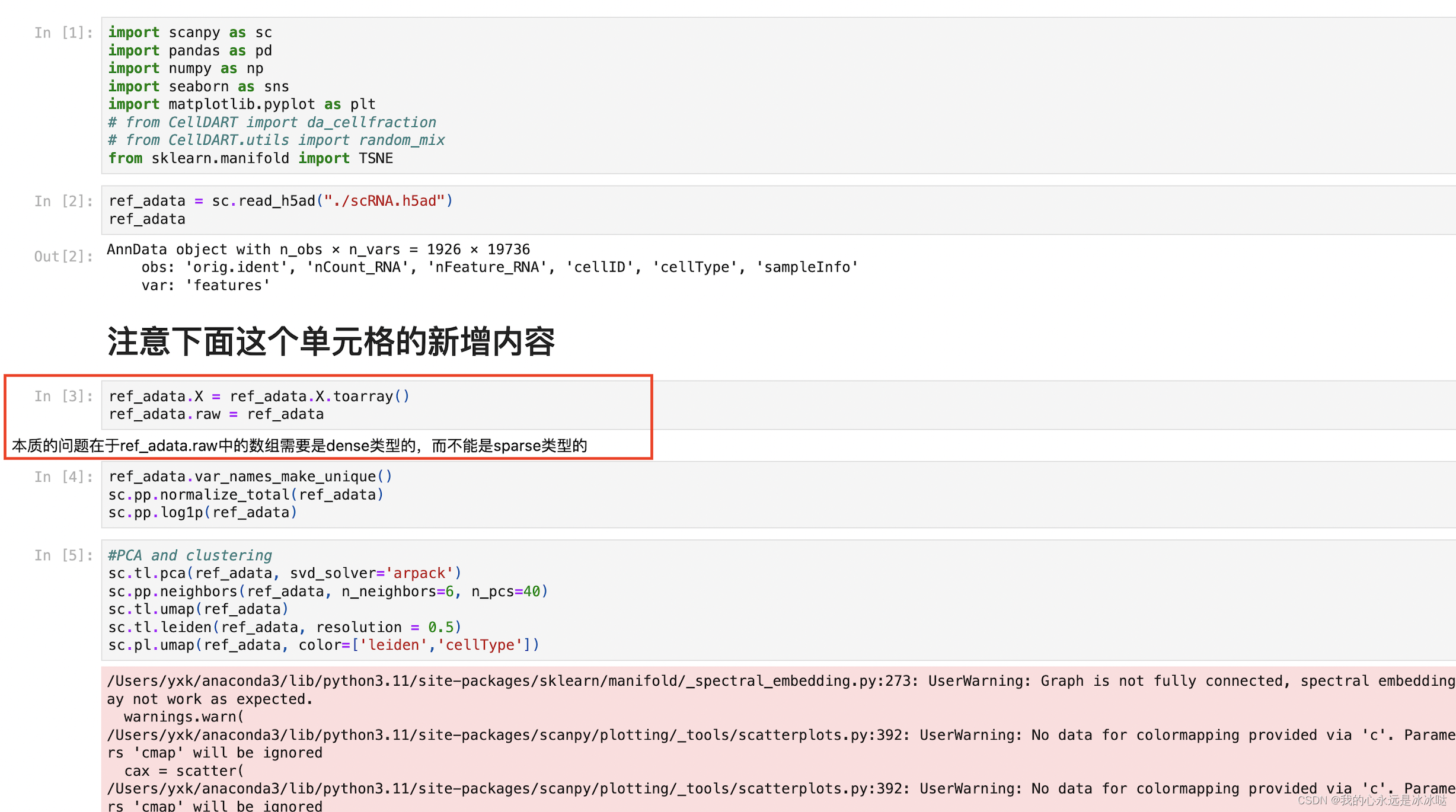
这个需要注意,有时间可以研究一下sc.tl.rank_genes_groups()内部是怎么实现的