前言
群体遗传学是研究生物群体中基因的分布、基因频率和基因型频率的维持和变化的学科。它不仅探讨遗传病的发病频率和遗传方式,还研究基因频率和变化的规律,为预防、监测和治疗遗传病提供重要信息。R语言作为一种强大的统计分析工具,在群体遗传学研究中扮演着不可或缺的角色。它提供了多种包和函数,使得复杂的遗传数据分析变得简单高效。本教程参考《Population genetics with R》
对于R语言的基础,我写过一些博客,可以先看R语言入门的教学
【R语言从0到精通】-1-下载R语言与R最基础内容-CSDN博客
R语言Matrix快速review
矩阵运算对于计算生物学的计算或者生物的计算来说是非常重要的。
现在你应该已经有配置好的Rstudio,然后熟悉基本的指令,那么现在我们学习一些学习本教程所需要的R语言小知识:对于Matrix操作及计算
1.Matrix建立
与数据框不同,R中的矩阵只接受相同类别的元素。因此,你不能拥有一个同时包含数字和因子(分类变量)的单一矩阵,只能包含其中一种。
R
# 创建一个3x3的矩阵,元素为1到9
mat <- matrix(1:9, nrow = 3, ncol = 3)
# 打印矩阵
print(mat)得到3*3矩阵:

如果我们使用byrow这个参数,会得到一个不一样的矩阵,因为是按x填充的:
R
mat2 <- matrix(1:9, nrow = 3, ncol = 3, byrow = T)
print(mat2)
2.R语言matrix简单乘法
我们创立一个小矩阵,进行自乘的矩阵运算
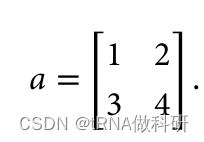
矩阵乘法的公式是:
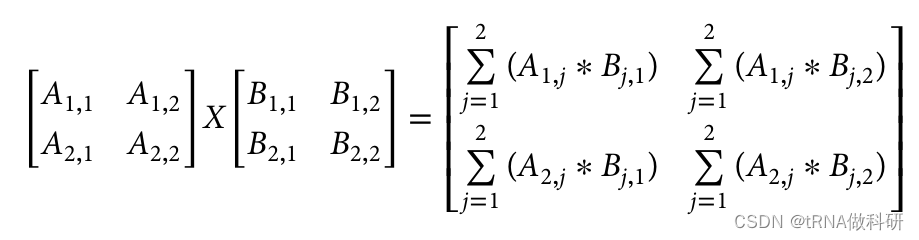
使用R语言得到:

而我们通过数学计算得到的是:
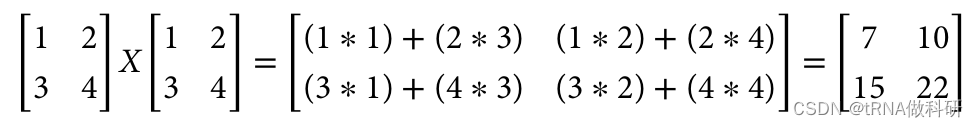
不一样的原因是R语言的矩阵乘法并不是简单的*,而是A %*% B
那我们在R语言中Matrix的*是描述什么运算呢?是对应位置的乘法,因此要求两个矩阵的行列数量是一样的,也就是:

好了,主要是为了讲一下矩阵的乘法在R语言中怎么用,接下来我们正式开始。
Hardy--Weinberg genotype proportions
1.一些名词
Allele: 等位基因:一种遗传变异(位于一对同源染色体相同位置上控制同一性状不同形态的基因)
Diploid: 二倍体:具有所有(大部分)遗传物质的两个副本的个体。
Haploid: 单倍体:具有所有遗传物质的一个副本的个体。
Heterozygote: 杂合子:具有不同等位基因副本的个体。
Homozygote: 纯合子:具有相同等位基因副本的个体。
Genotype: 基因型:个体中等位基因的组合。
Locus: 基因座:等位基因的物理位置,复数形式为loci。
Phenotype: 表型:基因型(和非遗传影响)的结果
2.群体遗传学简介
我将以通俗的语言讲述我对于群体遗传学的理解,希望大家能喜欢。
群体遗传学是一门研究群体中遗传信息传递和变化的科学。主要关注种群中遗传变异的模式,以及这些变异是如何随时间演变的。通俗地说,群体遗传学家就像是在玩一个大型的拼图游戏,我们试图弄清楚每个拼图片(即遗传变异或等位基因)在整个种群中的分布情况。
(1)首先,让我们来谈谈"预计有多少个体携带某一特定等位基因" 的问题。想象一下,我们有一盒五颜六色的积木,每种颜色的积木代表一种特定的等位基因。如果我们随机从盒子里拿出一块积木,我们可能会想知道拿出某种特定颜色积木的概率是多少。在群体遗传学中,这个概率可以通过查看该等位基因在种群中的频率来估计。如果一个等位基因在种群中很常见,那么携带它的个体数量就会相对较多;相反,如果它是罕见的,那么携带它的个体就会较少。
(2)"找到罕见突变两个副本的可能性有多大" 这个问题,可以比作是从积木盒子里连续拿出两块相同稀有颜色的积木的概率。因为罕见突变的等位基因在种群中出现的频率很低,所以同时拿到两个这样的副本的几率会非常小。这个概率可以通过计算该等位基因频率的平方来估计,因为每次复制事件都是独立的。
(3)"某一特定基因型来自一个人群或另一个人群的可能性有多大 "这个问题,可以想象成我们有一堆分别来自不同人群的积木 ,每堆积木中各种颜色(即基因型)的比例 可能不同。当我们看到一个特定颜色的积木时,我们可能会好奇它最有可能来自哪一堆。这个可能性可以通过比较不同人群中该基因型的频率 来确定。 有了这些基本的频率和概率预期之后,群体遗传学家就可以开始寻找实际观察到的遗传模式与预期之间的偏差。这些偏差可以提供有关种群历史、迁移事件、自然选择作用以及其他影响遗传变异分布的因素的重要线索。通过分析这些偏差,科学家们可以对种群的过去、现在和未来做出更加有根据的预测,就像是通过拼图的碎片逐渐揭示出整个画面的过程。
3.等位基因与SNP
等位基因在经典遗传学中被定义为导致不同表型的基因变体 ,可以是杂合子或纯合子。然而,在现代遗传学的语境中,等位基因的概念被扩展到包括基因组内特定位点或位置的任何遗传变异,不论这种变异是否对基因功能或生物体的表型产生影响。 一种特别普遍的遗传变异类型是单核苷酸多态性(SNP)。SNP发生在DNA序列中单个核苷酸的位置上,其中一个核苷酸在不同的个体中存在差异。例如,如果某个DNA序列的一部分原本是...GTAGCTAGAC. ..,而在另一个个体中这一部分变成了...GTAGTTAGAC ...这就构成了一个SNP。 当我们在多个个体间比较这段DNA序列时,会发现括号内的位置存在两种可能的核苷酸(C或T),这表明这个位点是一个多态性位点,拥有两个相关的等位基因。 在二倍体生物中,每个细胞包含两组染色体,因此每个基因位点上有两个等位基因。这意味着在考虑一个具有两个等位基因的位点时,可能存在三种不同的基因型:纯合子CC、杂合子CT和另一种纯合子TT。这种分类对于理解遗传变异如何在种群中分布以及它们如何可能影响生物体的性状至关重要。
4.等位基因频率
假设T等位基因在某个群体中频率为p(0-1)。当我们从这个群体中随机抽取一个等位基因时,抽到T等位基因的概率就是p。 如果T等位基因的频率是1/2,那么在一个没有额外亲本基因型信息的随机情况下,一个个体从一个亲本那里继承到这个T等位基因副本的概率也是1/2。这是因为每个亲本都有相等的机会传递T或非T等位基因 。 进一步考虑到新一代的个体,预计其中一半会从他们的一个亲本那里继承到T等位基因。由于每个亲本传递特定等位基因的概率独立,新一代中的某些个体还有可能从另一个亲本那里也继承到T等位基因,这个概率同样是1/2。 因此,在新一代的所有个体中,预计有一半的一半,即1/4的个体将会是两个T等位基因的纯合子 。这些纯合子的频率可以用数学表达式p × p = p^2 来表示。 这里隐含的假设是,这些遗传变异是按照随机的方式传递的。 在模拟随时间推移的群体遗传学时,我们需要做出许多这样的假设,以便简化模型并使问题可解。
5.Hardy--Weinberg genotype proportions
我们可以从观察到的基因型频率 反推预期的等位基因频率。一些遗传变异在纯合子时会产生隐性表型。假设我们在一个人群中观察到具有新的突变等位基因的纯合子,其频率为1/1000,也就是p方
R
f <- 1/1000
sqrt(f)接下来我们考虑杂合子的情况:
如果继承一个等位基因的概率是p,那么不继承它的概率就是1-p。因为我们在这里只关注杂合子的情况,这些个体要么从母亲那里继承了一个等位基因副本,但没有从父亲那里继承,概率为p(1-p),要么没有从母亲那里继承副本,但从父亲那里继承了,概率也为(1-p)p。这两个事件是互斥的:为了使个体成为携带者,突变等位基因要么来自母亲,要么来自父亲。因此,我们使用概率的"或"(加法)规则,将不同的结果相加:p(1-p) + (1-p)p = 2p(1-p)。
这些关于p2纯合子频率和2p(1-p)杂合子频率的预期被称为哈代-温伯格基因型比例。
我们可以通过R语言curve函数来可视化纯合子情况
R
curve(x^2, 0, 1,
xlab = "Allele Frequency", ylab = "Genotype Frequency", lwd = 3,col = "green")
text(0.6, 0.2, "Homozygotes",col = "green")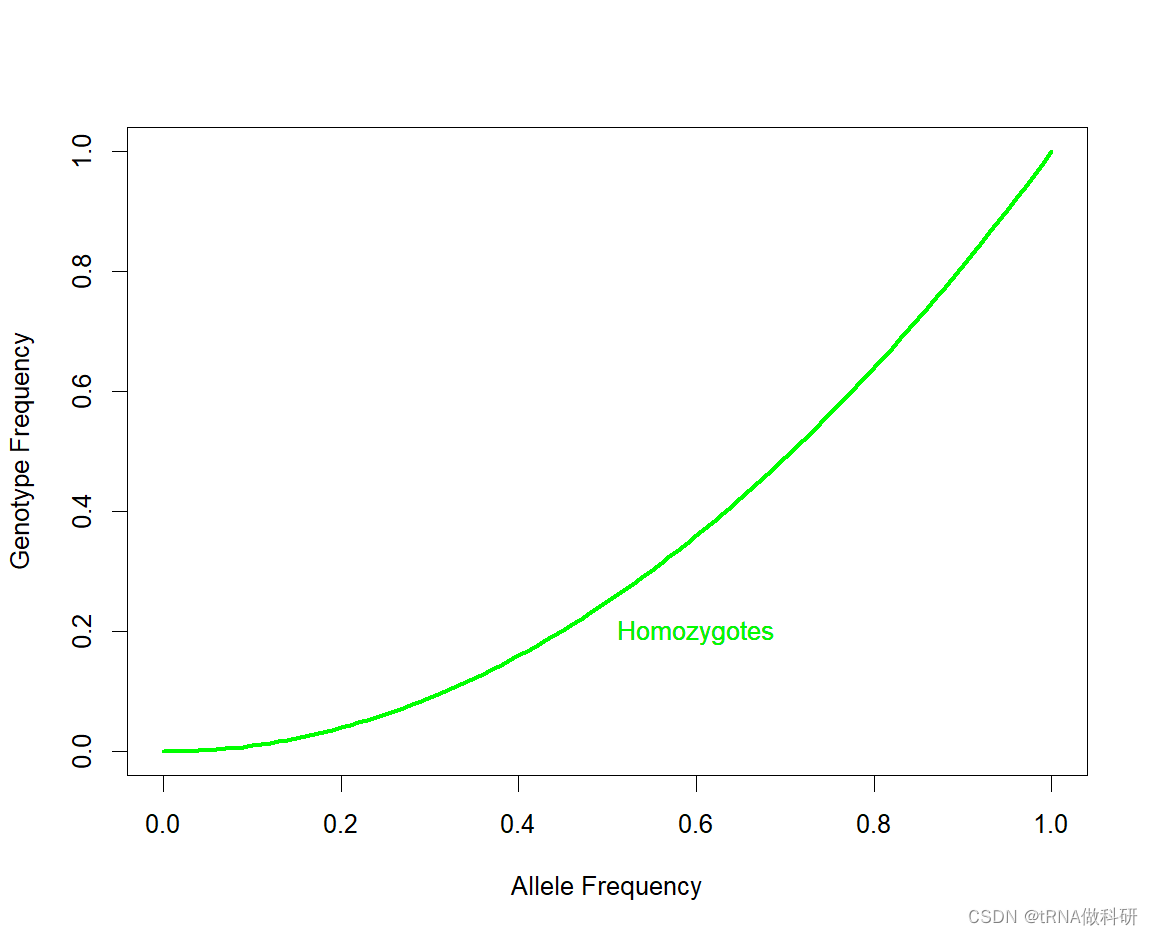
我们再加入杂合子的情况:
R
curve(2*x*(1-x), 0, 1, add=TRUE, xlab="Allele frequencies",
ylab="Genotype frequencies", col="blue", lwd=2)
text(0.25, 0.5, "Heterozygotes", col="blue")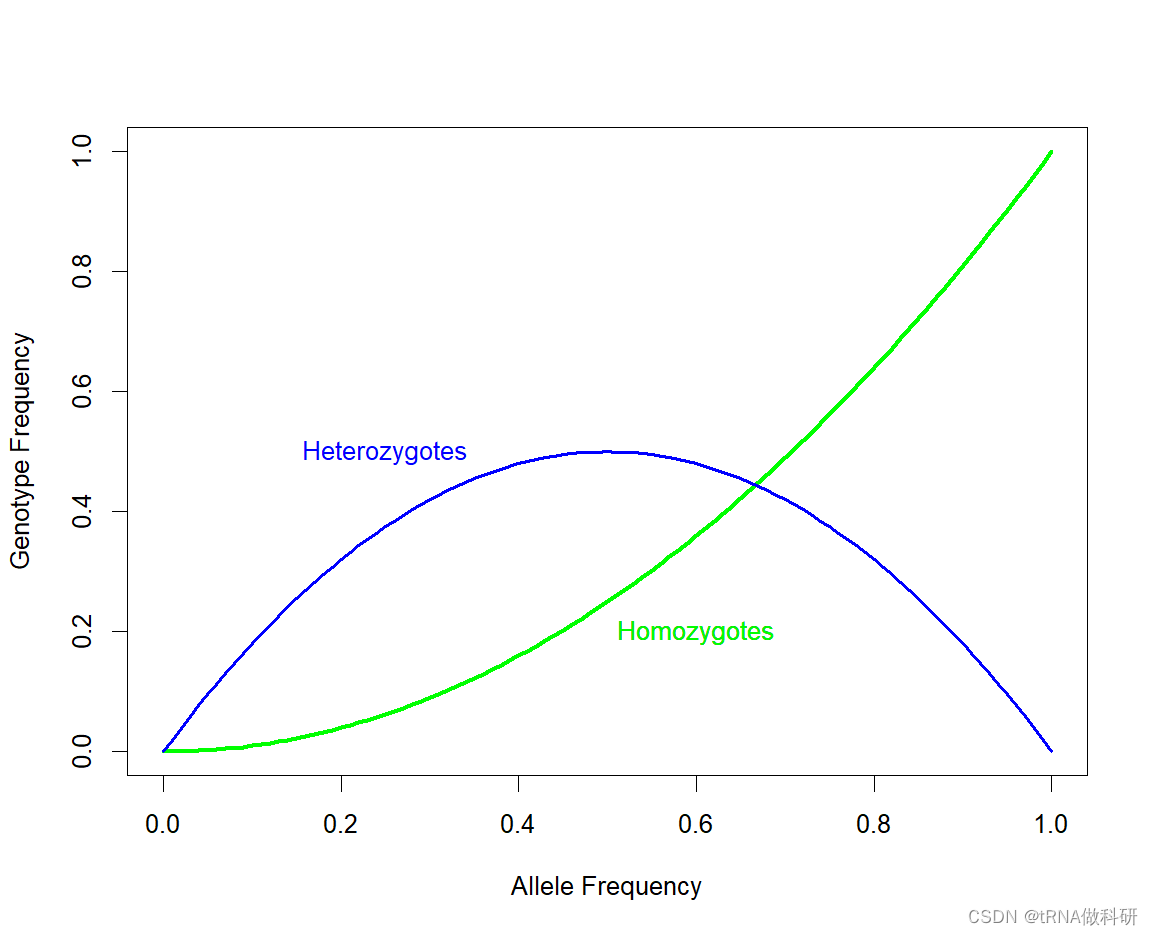
我们看到随着等位基因频率增加,杂合子和纯合子的频率预期不同。随等位基因频率的增加,杂合子的数量最初比纯合子的数量增长得更快。但是,当你的等位基因频率接近50%时,杂合子的数量趋于稳定,一旦大部分人口(超过50%)携带这个等位基因,更多的个体开始以纯合子的形式携带两份拷贝,而不是以杂合子的形式只携带一份拷贝 。当一个等位基因频率达到1.0(100%)时,这个等位基因被认为在种群中"固定"了。一旦发生这种情况,它只能存在于纯合子个体中,因为没有其他变异等位基因存在。当一个等位基因频率达到0%时,这个等位基因被认为是"丢失"了。
在图中我们可以看到一个两曲线相交的点,也就是: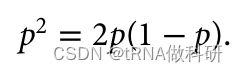
可以解得p=2/3。我们在图上标记:
R
p <- 2/3
points(p,p^2,lwd=2,cex=2)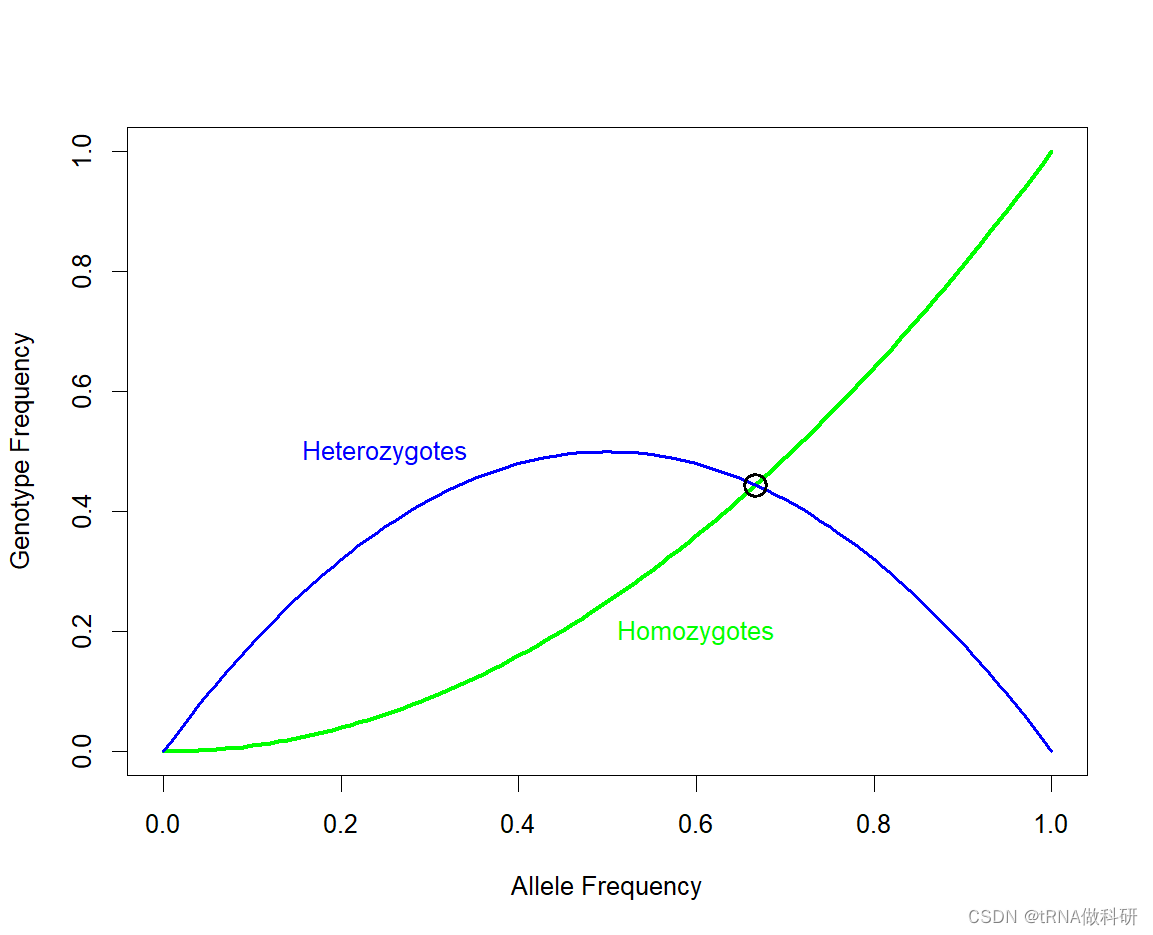
本次内容先写这么多,下一章讲simulateing genotype内容,欢迎大家在评论区讨论。