你可以将 RFdiffusion 理解为「蛋白质宇宙的建构者」。它由蛋白质设计领域的核心团队------David Baker 实验室于 2022 年提出,该框架以 RoseTTAFold 为骨架,引入扩散 生成模型 ,将结构预测与生成设计融合,首次实现了从头构建功能明确、构型复杂的蛋白质。 RFdiffusion 能够根据已有的功能核心自动补全蛋白质结构,生成高度可控的空间构型,并在多个蛋白质设计任务中展现出强大的通用性与创造性。
其功能覆盖六个关键方向:可根据指定基序构建骨架、从零生成全新蛋白质、设计具有对称性要求的结构、生成多样化的功能变体、精确设计结合界面,甚至可在具备对称约束的前提下完成复杂结构的补全。无论是基础研究、酶设计,还是分子对接与药物开发,RFdiffusion 都提供了一个强有力的生成式解决方案。
使用云平台: OpenBayes
首先点击「公共教程」,在公共教程中找到「RFdiffusion:扩散式蛋白设计模型」,单击打开。
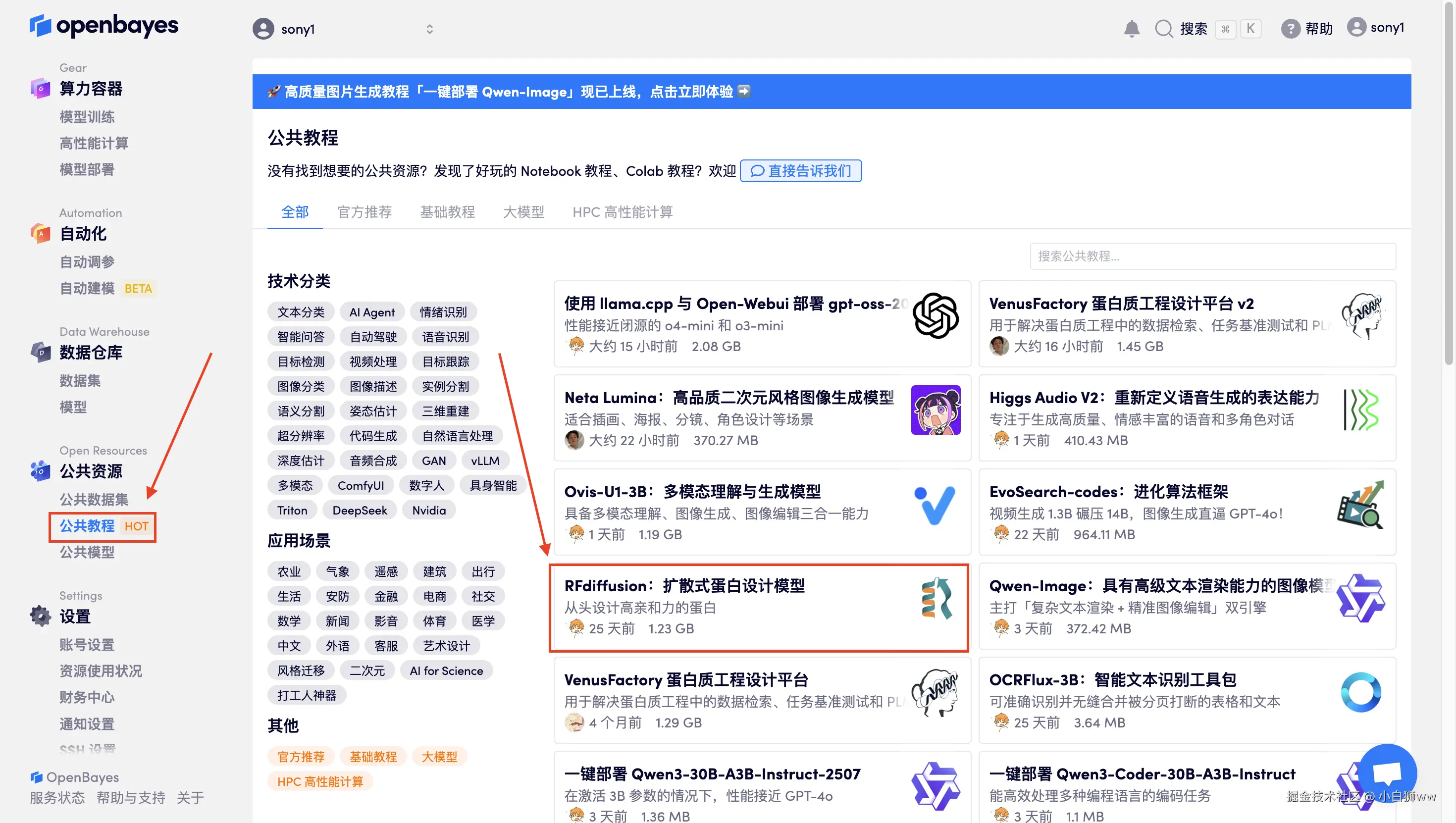
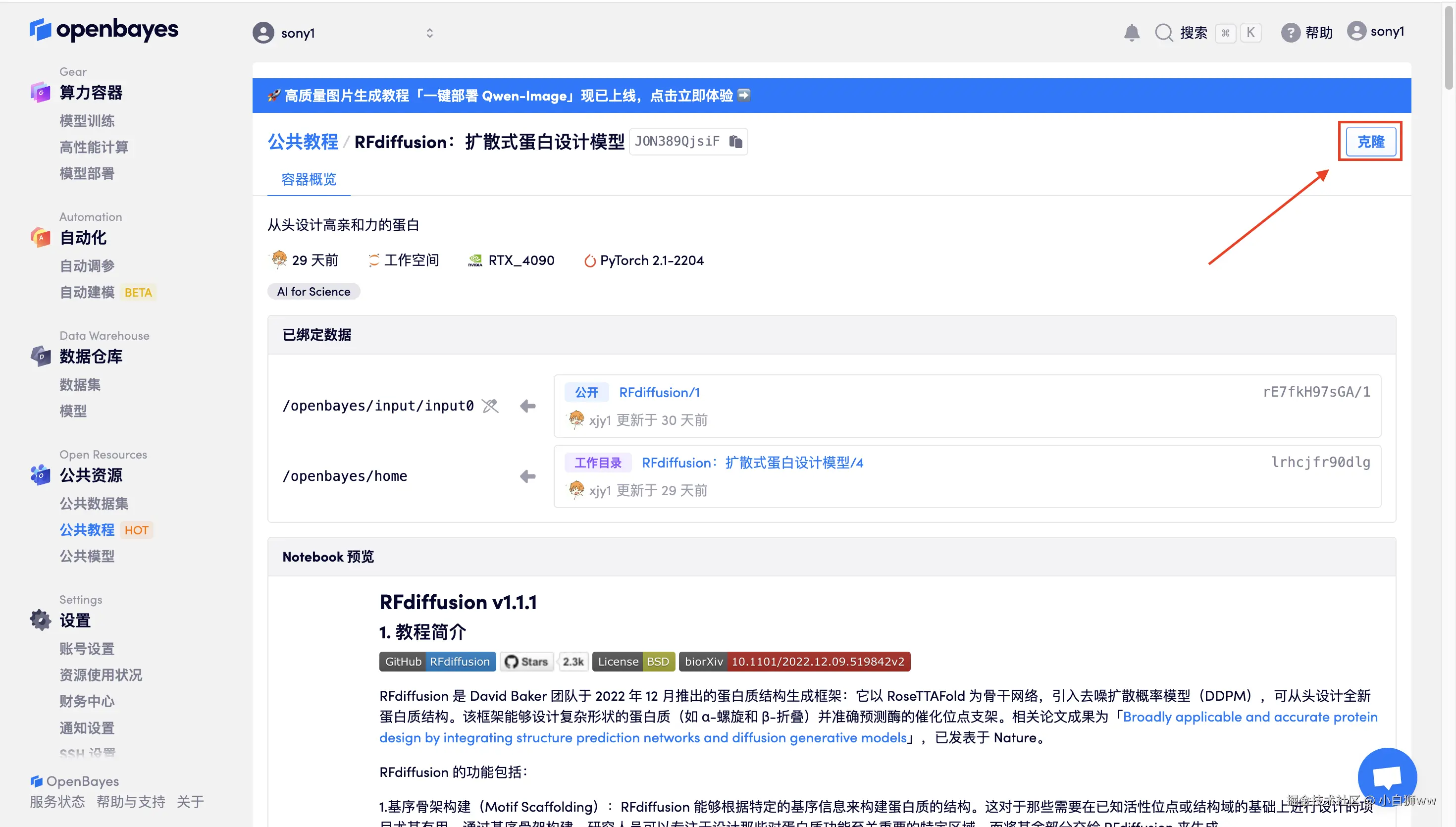
页面跳转后,点击右上角「克隆」,将该教程克隆至自己的容器中。
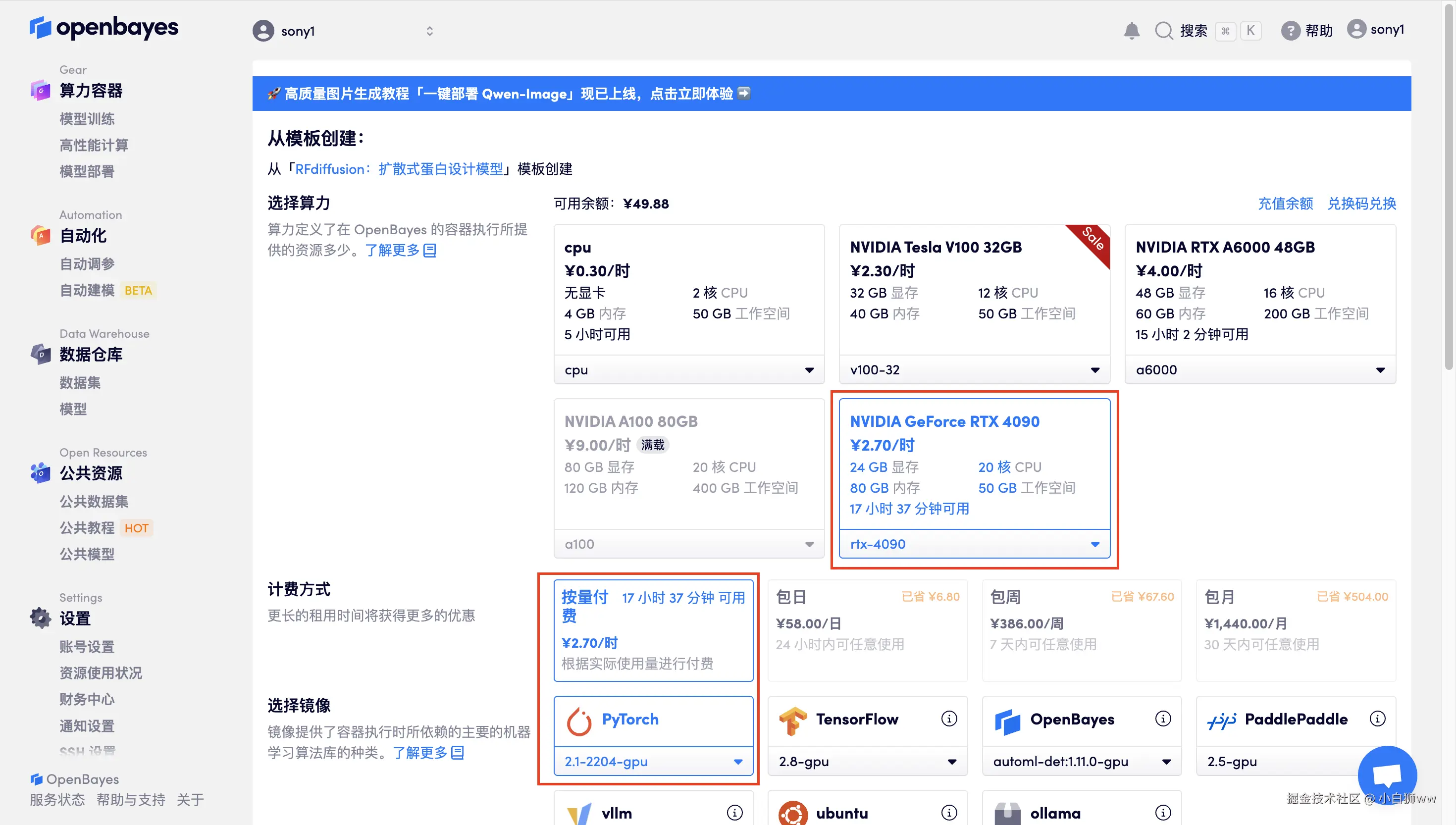
在当前页面中看到的算力资源均可以在平台一键选择使用。平台会默认选配好原教程所使用的算力资源、镜像版本,不需要再进行手动选择。点击「继续执行」,等待分配资源。
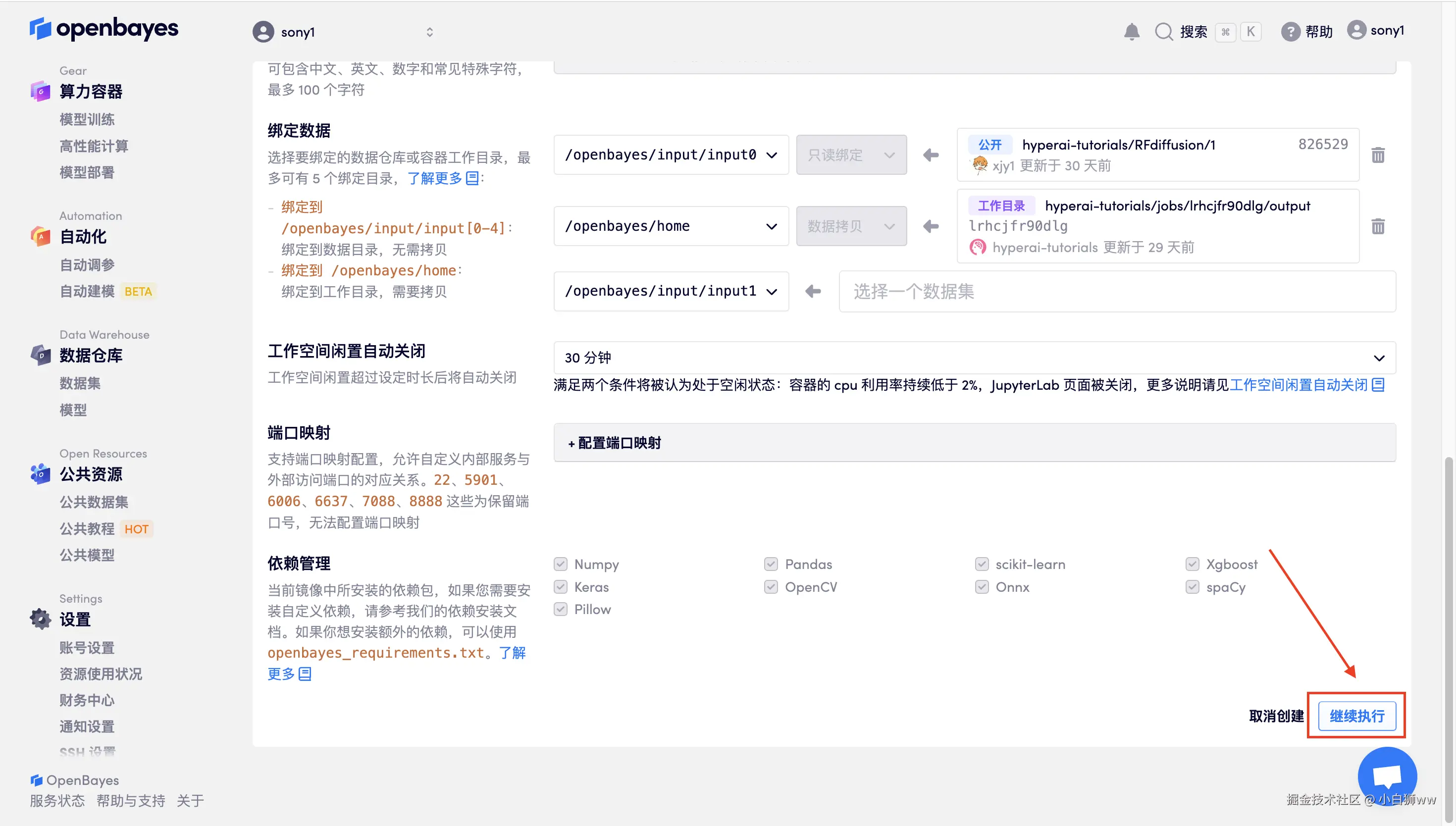
数据和代码都已经同步完成了。容器状态显示为「运行中」后,点击「打开工作空间」。
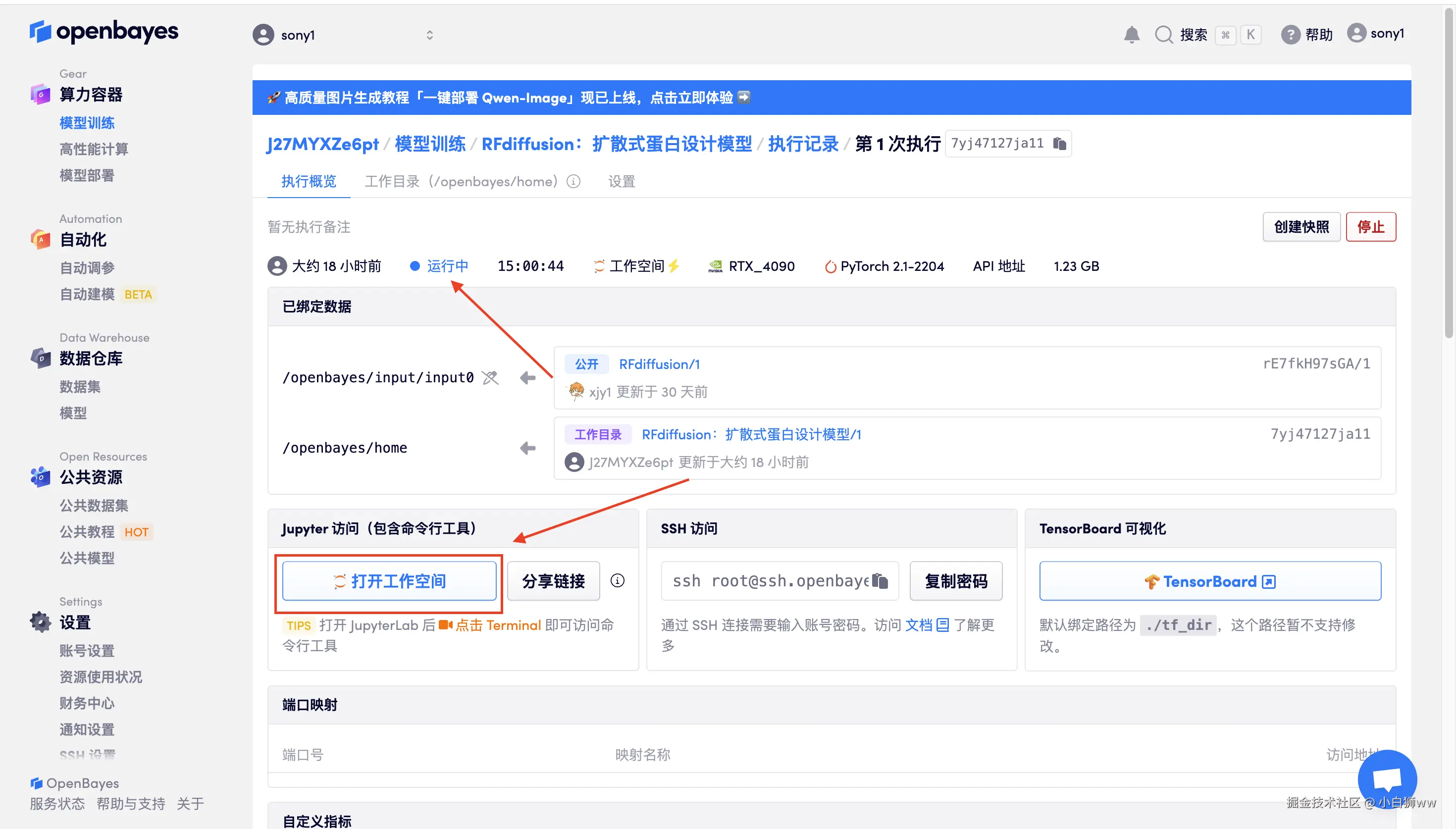
1.设置 RFdiffusion 扩散
python
import numpy as np
import os, time, signal
import sys, random, string, re
import zipfile
from IPython.display import display, HTML
if 'RFdiffusion' not in sys.path:
os.environ["DGLBACKEND"] = "pytorch"
sys.path.append('RFdiffusion')
os.environ["CUDA_VISIBLE_DEVICES"] = "0"
os.environ["MKL_THREADING_LAYER"] = "GNU"
import json
import matplotlib.pyplot as plt
from IPython.display import display, HTML
import ipywidgets as widgets
import py3Dmol
from inference.utils import parse_pdb
from colabdesign.rf.utils import get_ca
from colabdesign.rf.utils import fix_contigs, fix_partial_contigs, fix_pdb, sym_it
from colabdesign.shared.protein import pdb_to_string
from colabdesign.shared.plot import plot_pseudo_3D
from ipywidgets import FileUpload
from IPython.display import display
import subprocess
def get_pdb(pdb_code=None):
print("pdb_code",pdb_code)
if pdb_code is None or pdb_code == "":
uploader = FileUpload(description="Upload PDB", multiple=False)
display(uploader)
while not uploader.value:
time.sleep(0.1)
uploaded_filename = next(iter(uploader.value))
pdb_bytes = uploader.value[uploaded_filename]["content"]
with open("tmp.pdb", "wb") as out:
out.write(pdb_bytes)
return "tmp.pdb"
elif os.path.isfile(pdb_code):
return pdb_code
elif len(pdb_code) == 4:
if not os.path.isfile(f"{pdb_code}.pdb1"):
os.system(f"wget -qnc https://files.rcsb.org/download/{pdb_code}.pdb1.gz")
os.system(f"gunzip {pdb_code}.pdb1.gz")
return f"{pdb_code}.pdb1"
else:
os.system(f"wget -qnc https://alphafold.ebi.ac.uk/files/AF-{pdb_code}-F1-model_v3.pdb")
return f"AF-{pdb_code}-F1-model_v3.pdb"
def run_ananas(pdb_str, path, sym=None):
pdb_filename = f"outputs/{path}/ananas_input.pdb"
out_filename = f"outputs/{path}/ananas.json"
with open(pdb_filename, "w") as handle:
handle.write(pdb_str)
cmd = f"./ananas {pdb_filename} -u -j {out_filename}"
if sym is None:
os.system(cmd)
else:
os.system(f"{cmd} {sym}")
try:
out = json.loads(open(out_filename, "r").read())
results, AU = out[0], out[-1]["AU"]
group = AU["group"]
chains = AU["chain names"]
rmsd = results["Average_RMSD"]
print(f"AnAnaS detected {group} symmetry at RMSD:{rmsd:.3}")
C = np.array(results['transforms'][0]['CENTER'])
A = [np.array(t["AXIS"]) for t in results['transforms']]
new_lines = []
for line in pdb_str.split("\n"):
if line.startswith("ATOM"):
chain = line[21:22]
if chain in chains:
x = np.array([float(line[i:(i+8)]) for i in [30, 38, 46]])
if group[0] == "c":
x = sym_it(x, C, A[0])
if group[0] == "d":
x = sym_it(x, C, A[1], A[0])
coord_str = "".join(["{:8.3f}".format(a) for a in x])
new_lines.append(line[:30] + coord_str + line[54:])
else:
new_lines.append(line)
return results, "\n".join(new_lines)
except:
return None, pdb_str
def run(command, steps, num_designs=1, visual="none"):
def run_command_and_get_pid(command):
pid_file = '/dev/shm/pid'
os.system(f'nohup {command} > /dev/null & echo $! > {pid_file}')
with open(pid_file, 'r') as f:
pid = int(f.read().strip())
os.remove(pid_file)
return pid
def is_process_running(pid):
try:
os.kill(pid, 0)
except OSError:
return False
else:
return True
run_output = widgets.Output()
progress = widgets.FloatProgress(min=0, max=1, description='running', bar_style='info')
display(widgets.VBox([progress, run_output]))
for n in range(steps):
if os.path.isfile(f"/dev/shm/{n}.pdb"):
os.remove(f"/dev/shm/{n}.pdb")
pid = run_command_and_get_pid(command)
try:
fail = False
for _ in range(num_designs):
for n in range(steps):
wait = True
while wait and not fail:
time.sleep(0.1)
if os.path.isfile(f"/dev/shm/{n}.pdb"):
pdb_str = open(f"/dev/shm/{n}.pdb").read()
if pdb_str[-3:] == "TER":
wait = False
elif not is_process_running(pid):
fail = True
elif not is_process_running(pid):
fail = True
if fail:
progress.bar_style = 'danger'
progress.description = "failed"
break
else:
progress.value = (n+1) / steps
if visual != "none":
with run_output:
run_output.clear_output(wait=True)
if visual == "image":
xyz, bfact = get_ca(f"/dev/shm/{n}.pdb", get_bfact=True)
fig = plt.figure()
fig.set_dpi(100);fig.set_figwidth(6);fig.set_figheight(6)
ax1 = fig.add_subplot(111);ax1.set_xticks([]);ax1.set_yticks([])
plot_pseudo_3D(xyz, c=bfact, cmin=0.5, cmax=0.9, ax=ax1)
plt.show()
if visual == "interactive":
view = py3Dmol.view(js='https://3dmol.org/build/3Dmol.js')
view.addModel(pdb_str, 'pdb')
view.setStyle({'cartoon': {'colorscheme': {'prop': 'b', 'gradient': 'roygb', 'min': 0.5, 'max': 0.9}}})
view.zoomTo()
view.show()
if os.path.exists(f"/dev/shm/{n}.pdb"):
os.remove(f"/dev/shm/{n}.pdb")
if fail:
progress.bar_style = 'danger'
progress.description = "failed"
break
while is_process_running(pid):
time.sleep(0.1)
except KeyboardInterrupt:
os.kill(pid, signal.SIGTERM)
progress.bar_style = 'danger'
progress.description = "stopped"
def run_diffusion(contigs, path, pdb=None, iterations=50,
symmetry="none", order=1, hotspot=None,
chains=None, add_potential=False,
num_designs=1, visual="none"):
full_path = f"outputs/{path}"
os.makedirs(full_path, exist_ok=True)
opts = [f"inference.output_prefix={full_path}",
f"inference.num_designs={num_designs}"]
if chains == "":
chains = None
if symmetry in ["auto", "cyclic", "dihedral"]:
if symmetry == "auto":
sym, copies = None, 1
else:
sym, copies = {"cyclic": (f"c{order}", order),
"dihedral": (f"d{order}", order * 2)}[symmetry]
else:
symmetry = None
sym, copies = None, 1
contigs = contigs.replace(",", " ").replace(":", " ").split()
is_fixed, is_free = False, False
fixed_chains = []
for contig in contigs:
for x in contig.split("/"):
a = x.split("-")[0]
if a[0].isalpha():
is_fixed = True
if a[0] not in fixed_chains:
fixed_chains.append(a[0])
if a.isnumeric():
is_free = True
if len(contigs) == 0 or not is_free:
mode = "partial"
elif is_fixed:
mode = "fixed"
else:
mode = "free"
if mode in ["partial", "fixed"]:
pdb_str = pdb_to_string(get_pdb(pdb), chains=chains)
if symmetry == "auto":
a, pdb_str = run_ananas(pdb_str, path)
if a is None:
print(f'ERROR: no symmetry detected')
symmetry = None
sym, copies = None, 1
else:
if a["group"][0] == "c":
symmetry = "cyclic"
sym, copies = a["group"], int(a["group"][1:])
elif a["group"][0] == "d":
symmetry = "dihedral"
sym, copies = a["group"], 2 * int(a["group"][1:])
else:
print(f'ERROR: the detected symmetry ({a["group"]}) not currently supported')
symmetry = None
sym, copies = None, 1
elif mode == "fixed":
pdb_str = pdb_to_string(pdb_str, chains=fixed_chains)
pdb_filename = f"{full_path}/input.pdb"
with open(pdb_filename, "w") as handle:
handle.write(pdb_str)
parsed_pdb = parse_pdb(pdb_filename)
opts.append(f"inference.input_pdb={pdb_filename}")
if mode in ["partial"]:
iterations = int(80 * (iterations / 200))
opts.append(f"diffuser.partial_T={iterations}")
contigs = fix_partial_contigs(contigs, parsed_pdb)
else:
opts.append(f"diffuser.T={iterations}")
contigs = fix_contigs(contigs, parsed_pdb)
else:
opts.append(f"diffuser.T={iterations}")
parsed_pdb = None
contigs = fix_contigs(contigs, parsed_pdb)
if hotspot is not None and hotspot != "":
opts.append(f"ppi.hotspot_res=[{hotspot}]")
if sym is not None:
sym_opts = ["--config-name symmetry", f"inference.symmetry={sym}"]
if add_potential:
sym_opts += ["'potentials.guiding_potentials=["type:olig_contacts,weight_intra:1,weight_inter:0.1"]'",
"potentials.olig_intra_all=True", "potentials.olig_inter_all=True",
"potentials.guide_scale=2", "potentials.guide_decay=quadratic"]
opts = sym_opts + opts
contigs = sum([contigs] * copies, [])
opts.append(f"'contigmap.contigs=[{' '.join(contigs)}]'")
opts += ["inference.dump_pdb=True", "inference.dump_pdb_path='/dev/shm'"]
print("mode:", mode)
print("output:", full_path)
print("contigs:", contigs)
opts_str = " ".join(opts)
print(opts_str)
cmd =f"bash -c 'source activate /openbayes/input/input0/py3102 && python /openbayes/home/RFdiffusion/run_inference.py {opts_str}'"
print(cmd)
run(cmd, iterations, num_designs, visual=visual)
print("-"*20)
for n in range(num_designs):
pdbs = [f"/openbayes/home/outputs/traj/{path}_{n}_pX0_traj.pdb",
f"/openbayes/home/outputs/traj/{path}_{n}_Xt-1_traj.pdb",
f"{full_path}_{n}.pdb"]
for pdb in pdbs:
with open(pdb, "r") as handle:
pdb_str = handle.read()
with open(pdb, "w") as handle:
handle.write(fix_pdb(pdb_str, contigs))
return contigs, copies
less
/openbayes/input/input0/py3102/lib/python3.10/site-packages/requests/__init__.py:86: RequestsDependencyWarning: Unable to find acceptable character detection dependency (chardet or charset_normalizer).
warnings.warn(
/output/RFdiffusion/Track_module.py:241: FutureWarning: `torch.cuda.amp.autocast(args...)` is deprecated. Please use `torch.amp.autocast('cuda', args...)` instead.
@torch.cuda.amp.autocast(enabled=False)2.运行 RFdiffusion 以生成 backbone
ruby
#@title run **RFdiffusion** to generate a backbone
name = "test" #@param {type:"string"}
contigs = "100" #@param {type:"string"}
pdb = "" #@param {type:"string"}
iterations = 25 #@param [25, 50, 100, 150, 200] {type:"raw"}
hotspot = "" #@param {type:"string"}
num_designs = 1 #@param [1, 2, 4, 8, 16, 32] {type:"raw"}
visual = "interactive" #@param ["none", "image", "interactive"]
#@markdown ---
#@markdown **symmetry** settings
#@markdown ---
symmetry = "auto" #@param ["none", "auto", "cyclic", "dihedral"]
order = 1 #@param [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12] {type:"raw"}
chains = "" #@param {type:"string"}
add_potential = True #@param {type:"boolean"}
#@markdown - `symmetry='auto'` enables automatic symmetry dectection with [AnAnaS](https://team.inria.fr/nano-d/software/ananas/).
#@markdown - `chains="A,B"` filter PDB input to these chains (may help auto-symm detector)
#@markdown - `add_potential` to discourage clashes between chains
path = name
while os.path.exists(f"/openbayes/home/outputs/{path}_0.pdb"):
path = name + "_" + ''.join(random.choices(string.ascii_lowercase + string.digits, k=5))
flags = {"contigs":contigs,
"pdb":pdb,
"order":order,
"iterations":iterations,
"symmetry":symmetry,
"hotspot":hotspot,
"path":path,
"chains":chains,
"add_potential":add_potential,
"num_designs":num_designs,
"visual":visual}
for k,v in flags.items():
if isinstance(v,str):
flags[k] = v.replace("'","").replace('"','')
contigs, copies = run_diffusion(**flags)
ini
mode: free
output: outputs/test_s67hh
contigs: ['100-100']
inference.output_prefix=outputs/test_s67hh inference.num_designs=1 diffuser.T=25 'contigmap.contigs=[100-100]' inference.dump_pdb=True inference.dump_pdb_path='/dev/shm'
bash -c 'source activate /openbayes/input/input0/py3102 && python /openbayes/home/RFdiffusion/run_inference.py inference.output_prefix=outputs/test_s67hh inference.num_designs=1 diffuser.T=25 'contigmap.contigs=[100-100]' inference.dump_pdb=True inference.dump_pdb_path='/dev/shm''
ini
VBox(children=(FloatProgress(value=0.0, bar_style='info', description='running', max=1.0), Output()))
less
/openbayes/input/input0/py3102/lib/python3.10/site-packages/requests/__init__.py:86: RequestsDependencyWarning: Unable to find acceptable character detection dependency (chardet or charset_normalizer).
warnings.warn(
/output/RFdiffusion/Track_module.py:241: FutureWarning: `torch.cuda.amp.autocast(args...)` is deprecated. Please use `torch.amp.autocast('cuda', args...)` instead.
@torch.cuda.amp.autocast(enabled=False)
/output/RFdiffusion/inference/model_runners.py:175: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
self.ckpt = torch.load(
/output/RFdiffusion/util_module.py:259: UserWarning: Using torch.cross without specifying the dim arg is deprecated.
Please either pass the dim explicitly or simply use torch.linalg.cross.
The default value of dim will change to agree with that of linalg.cross in a future release. (Triggered internally at ../aten/src/ATen/native/Cross.cpp:62.)
CBrotaxis1 = (CBr-CAr).cross(NCr-CAr)参数说明:
-
name:设计名称前缀,用于标识生成的输出文件。
-
contigs:指定要生成的蛋白质长度(残基数)。
-
pdb:输入参考的 PDB 文件路径(可选),如果提供,将基于该结构进行设计。
-
iterations:扩散过程的迭代次数。值越大生成质量可能越高,但计算时间越长。
-
hotspot:指定关键残基位置(热点残基),用于约束特定位置的氨基酸类型。
-
num_designs:要生成的设计数量,生成多个设计时可进行筛选。
-
visual:结果可视化方式。
- none:不生成可视化。
- image:生成静态图片。
- interactive:生成交互式 3D 可视化。
-
symmetry:对称性类型。
- none:无对称性。
- auto:自动检测对称性(使用 AnAnaS 算法)。
- cyclic:循环对称。
- dihedral:二面体对称。
-
order:对称阶数(亚基数量)。
-
chains:指定 PDB 文件中使用的链。
-
add_potential:是否添加额外势能防止链间碰撞。参数说明:
-
name:设计名称前缀,用于标识生成的输出文件。
-
contigs:指定要生成的蛋白质长度(残基数)。
-
pdb:输入参考的 PDB 文件路径(可选),如果提供,将基于该结构进行设计。
-
iterations:扩散过程的迭代次数。值越大生成质量可能越高,但计算时间越长。
-
hotspot:指定关键残基位置(热点残基),用于约束特定位置的氨基酸类型。
-
num_designs:要生成的设计数量,生成多个设计时可进行筛选。
-
visual:结果可视化方式。
- none:不生成可视化。
- image:生成静态图片。
- interactive:生成交互式 3D 可视化。
-
symmetry:对称性类型。
- none:无对称性。
- auto:自动检测对称性(使用 AnAnaS 算法)。
- cyclic:循环对称。
- dihedral:二面体对称。
-
order:对称阶数(亚基数量)。
-
chains:指定 PDB 文件中使用的链。
-
add_potential:是否添加额外势能防止链间碰撞。
基本语法
- 使用
contigs定义连续链。 - 使用
:来分隔多个连续链(contig),使用/在一个连续链内定义多个片段(segment)。
示例说明
-
无条件设计(Unconditional)
contigs=``'100'- 扩散一个长度为 100 的单体(monomer)。contigs=``'50:100'- 扩散一个长度为 50 和 100 的异源寡聚体(hetero-oligomer)。contigs=``'50'且设置symmetry=``'cyclic'和order=2- 将定义的连续链复制两份,并添加对称性约束,用于同源寡聚体(homo-oligomeric)扩散。
-
结合蛋白设计(Binder Design)
contigs=``'A:50'且设置pdb=``'4N5T'- 扩散一个长度为 50 的结合蛋白(binder),靶向指定 PDB 文件中的链 A。contigs=``'E6-155:70-100'且设置pdb=``'5KQV'和hotspot=``'E64,E88,E96'- 扩散一个长度在 70 到 100 之间(随机采样)的结合蛋白,靶向链 E,并指定热点残基(hotspots)。
-
基序支架(Motif Scaffolding)
contigs=``'40/A163-181/40'且设置pdb=``'5TPN'- 在指定 PDB 片段(A163-181)的两端各扩散 40 个残基。contigs=``'A3-30/36/A33-68'且设置pdb=``'6MRR'- 在两个指定的 PDB 片段(A3-30和A33-68)之间扩散一段长度为 36 的环(loop)。
-
部分扩散(Partial Diffusion)
contigs=``''且设置pdb=``'6MRR'- 对PDB中所有坐标添加噪声(即整个结构都参与扩散)。contigs=``'A1-10'且设置pdb=``'6MRR'- 固定前 10 个残基,其余部分添加噪声(即只对指定范围外的部分扩散)。contigs=``'A'且设置pdb=``'1SSC'- 固定链 A,对其它链添加噪声(即只对非指定链扩散)。
-
提示与技巧(Hints and Tips)
pdb=``''留空,程序会提示上传 PDB 文件。contigs=``'50-100'使用连字符指定一个长度范围,程序会从中随机采样一个长度。
3.显示 3D 结构
css
#@title Display 3D structure {run: "auto"}
animate = "none" #@param ["none", "movie", "interactive"]
color = "chain" #@param ["rainbow", "chain", "plddt"]
denoise = True
dpi = 100 #@param [100, 200, 400] {type:"raw"}
from colabdesign.shared.plot import pymol_color_list
from colabdesign.rf.utils import get_ca, get_Ls, make_animation
from string import ascii_uppercase, ascii_lowercase
alphabet_list = list(ascii_uppercase + ascii_lowercase)
def plot_pdb(num=0):
if denoise:
pdb_traj = f"/openbayes/home/outputs/traj/{path}_{num}_pX0_traj.pdb"
else:
pdb_traj = f"/openbayes/home/outputs/traj/{path}_{num}_Xt-1_traj.pdb"
if animate in ["none", "interactive"]:
hbondCutoff = 4.0
view = py3Dmol.view(js='https://3dmol.org/build/3Dmol.js')
# view = py3Dmol.view(js='/openbayes/home/RFdiffusion/3Dmol.js')
if animate == "interactive":
pdb_str = open(pdb_traj, 'r').read()
view.addModelsAsFrames(pdb_str, 'pdb', {'hbondCutoff': hbondCutoff})
else:
pdb = f"outputs/{path}_{num}.pdb"
pdb_str = open(pdb, 'r').read()
view.addModel(pdb_str, 'pdb', {'hbondCutoff': hbondCutoff})
if color == "rainbow":
view.setStyle({'cartoon': {'color': 'spectrum'}})
elif color == "chain":
for n, chain, c in zip(range(len(contigs)),
alphabet_list,
pymol_color_list):
view.setStyle({'chain': chain}, {'cartoon': {'color': c}})
else:
view.setStyle({'cartoon': {'colorscheme': {'prop': 'b', 'gradient': 'roygb', 'min': 0.5, 'max': 0.9}}})
view.zoomTo()
if animate == "interactive":
view.animate({'loop': 'backAndForth'})
view.show()
else:
Ls = get_Ls(contigs)
xyz, bfact = get_ca(pdb_traj, get_bfact=True)
xyz = xyz.reshape((-1, sum(Ls), 3))[::-1]
bfact = bfact.reshape((-1, sum(Ls)))[::-1]
if color == "chain":
display(HTML(make_animation(xyz, Ls=Ls, dpi=dpi, ref=-1)))
elif color == "rainbow":
display(HTML(make_animation(xyz, dpi=dpi, ref=-1)))
else:
display(HTML(make_animation(xyz, plddt=bfact*100, dpi=dpi, ref=-1)))
if num_designs > 1:
output = widgets.Output()
def on_change(change):
if change['name'] == 'value':
with output:
output.clear_output(wait=True)
plot_pdb(change['new'])
dropdown = widgets.Dropdown(
options=[(f'{k}', k) for k in range(num_designs)],
value=0, description='design:',
)
dropdown.observe(on_change)
display(widgets.VBox([dropdown, output]))
with output:
plot_pdb(dropdown.value)
else:
plot_pdb()
参数说明:
-
animate:控制蛋白质结构的动态展示方式。
- none:静态显示最终结构(无动画)。
- movie:生成蛋白质折叠过程的动态电影。
- interactive:创建可交互的动画(可前后播放)。
-
color:决定蛋白质结构的着色方式。
- rainbow:彩虹色渐变(从 N 端到 C 端)。
- chain:按蛋白质链着色(每条链不同颜色)。
- plddt:根据预测置信度着色(蓝色 = 高置信度,红色 = 低置信度)。
-
denoise:轨迹选择。
- True:使用去噪轨迹(最终优化结构)。
- False:使用含噪声的轨迹(中间过程)。
4.运行 ProteinMPNN 以生成序列,然后使用 AlphaFold 进行验证。
ini
#@title run **ProteinMPNN** to generate a sequence and **AlphaFold** to validate
num_seqs = 8 #@param [1, 2, 4, 8, 16, 32, 64] {type:"raw"}
initial_guess = False #@param {type:"boolean"}
num_recycles = 1 #@param [0, 1, 2, 3, 6, 12] {type:"raw"}
use_multimer = False #@param {type:"boolean"}
rm_aa = "C" #@param {type:"string"}
mpnn_sampling_temp = 0.1 #@param [0.0001, 0.1, 0.15, 0.2, 0.25, 0.3, 0.5, 1.0] {type:"raw"}
#@markdown - for **binder** design, we recommend `initial_guess=True num_recycles=3`
import subprocess
if not os.path.isfile("/openbayes/input/input0/params/done.txt"):
print("downloading AlphaFold params...")
while not os.path.isfile("/openbayes/input/input0/params/done.txt"):
time.sleep(5)
contigs_str = ":".join(contigs)
opts = [f"--pdb=/openbayes/home/outputs/{path}_0.pdb",
f"--loc=/openbayes/home/outputs/{path}",
f"--contig={contigs_str}",
f"--copies={copies}",
f"--num_seqs={num_seqs}",
f"--num_recycles={num_recycles}",
f"--rm_aa={rm_aa}",
f"--mpnn_sampling_temp={mpnn_sampling_temp}",
f"--num_designs={num_designs}"]
if initial_guess: opts.append("--initial_guess")
if use_multimer: opts.append("--use_multimer")
opts = ' '.join(opts)
conda_prefix = "/openbayes/input/input0/py3102"
env = os.environ.copy()
env["LD_LIBRARY_PATH"] = f"{conda_prefix}/lib:" + env.get("LD_LIBRARY_PATH", "")
cmd_list = [
f"{conda_prefix}/bin/python",
"/openbayes/home/colabdesign/rf/designability_test.py",
] + opts.split()
print("Running:", " ".join(cmd_list))
subprocess.run(cmd_list, env=env, check=True)
ruby
Running: /openbayes/input/input0/py3102/bin/python /openbayes/home/colabdesign/rf/designability_test.py --pdb=/openbayes/home/outputs/test_s67hh_0.pdb --loc=/openbayes/home/outputs/test_s67hh --contig=100-100 --copies=1 --num_seqs=8 --num_recycles=1 --rm_aa=C --mpnn_sampling_temp=0.1 --num_designs=1
{'pdb':'/openbayes/home/outputs/test_s67hh_0.pdb','loc':'/openbayes/home/outputs/test_s67hh','contigs':'100-100','copies':1,'num_seqs':8,'initial_guess':False,'use_multimer':False,'num_recycles':1,'rm_aa':'C','num_designs':1,'mpnn_sampling_temp':0.1}
protocol=fixbb
running proteinMPNN...
running AlphaFold...
design:0 n:0 mpnn:1.038 plddt:0.909 ptm:0.754 pae:4.021 rmsd:0.605 KEKELKERIEKKIKAFGKELGKTSEKFFEFFKALLELVRKKGYEEVKKLLEEGPEALAKALKEELGVDISVSFIKSISKEELEKILEKAKEIVEEEKELE
design:0 n:1 mpnn:1.117 plddt:0.891 ptm:0.756 pae:4.604 rmsd:0.886 KEEELEKEIEERIKEFAKELGLTSEEFLELFRAILELVRKLGYEEVRRLLEEGPEALARALEEVLGKRVSVAFIRSLSRETLEKILEEAERIVEEEEKKK
design:0 n:2 mpnn:1.078 plddt:0.915 ptm:0.770 pae:3.792 rmsd:0.497 KEEEIEKKIEEKIKKFAEELGKTSEKFIELLKNILELVKKEGYEKVEELLKKGNEALAKALEEVLGAKISVKFLESISKEEKEKMLEHAKEIVEEEEELK
design:0 n:3 mpnn:1.052 plddt:0.894 ptm:0.726 pae:4.354 rmsd:0.859 KKEEIEKRIEEKIKKKAEELGKTSEEFIEIFKAIYELVKKKGYEEVRKLLEEGPEALAKALEEELGVKVKVSTLKSISKEEWEKILEFAKEIVEEEKELK
design:0 n:4 mpnn:1.034 plddt:0.903 ptm:0.757 pae:4.178 rmsd:0.574 KKEELEKRIEEKIKKFAKELGRTSPEFLELLKAIYELVKKKGYEEVEKLLKEGAEALAKALKEELGLDVPVSFIESISPEELEKMLKKAKEIVEEEKKLE
design:0 n:5 mpnn:1.136 plddt:0.908 ptm:0.784 pae:3.463 rmsd:0.647 AAAAVAEARDAAIRAFGAELGRTSPEFLTLARALLALVERLGYAEVRRLLEAGRAALAAALARELGLKVPVSFLESISPEELAALLEHAEALVAELRALA
design:0 n:6 mpnn:1.137 plddt:0.913 ptm:0.780 pae:3.729 rmsd:0.593 EEAALEEEVEERIRAFAEELGLTSPRFLELFRAILELVRRLGREEVRALLAAGAEALAAALKEVLGLDVPVSFLESLSPETWEAILEKAEEIAEELEERE
design:0 n:7 mpnn:1.070 plddt:0.946 ptm:0.841 pae:2.574 rmsd:0.721 SAAALEAAVEADIRAFGASLGLTSPAALAFFRALLALVRREGAAAVRALLAAGPEALAAALRERLGADVPVAFLRSLSPATLEAALAHAEALVAREAAAA
ini
CompletedProcess(args=['/openbayes/input/input0/py3102/bin/python', '/openbayes/home/colabdesign/rf/designability_test.py', '--pdb=/openbayes/home/outputs/test_s67hh_0.pdb', '--loc=/openbayes/home/outputs/test_s67hh', '--contig=100-100', '--copies=1', '--num_seqs=8', '--num_recycles=1', '--rm_aa=C', '--mpnn_sampling_temp=0.1', '--num_designs=1'], returncode=0)参数说明:
- num_seqs:指定为每个蛋白质骨架生成多少条候选序列
- initial_guess:是否使用 RFdiffusion 生成的骨架作为 AlphaFold 的初始结构
- num_recycles:控制 AlphaFold 的结构优化迭代次数
- use_multimer:是否使用 AlphaFold-Multimer 模型
- rm_aa:指定不在设计中使用的氨基酸类型
- mpnn_sampling_temp:控制 ProteinMPNN 序列生成的多样性/保守性
5.显示最佳结果
python
def plot_pdb(num="best"):
if num == "best":
with open(f"/openbayes/home/outputs/{path}/best.pdb", "r") as f:
info = f.readline().strip('\n').split()
num = info[3]
hbondCutoff = 4.0
view = py3Dmol.view(js='https://3dmol.org/build/3Dmol.js')
pdb_str = open(f"/openbayes/home/outputs/{path}_{num}.pdb", 'r').read()
view.addModel(pdb_str, 'pdb', {'hbondCutoff': hbondCutoff})
pdb_str = open(f"/openbayes/home/outputs/{path}/best_design{num}.pdb", 'r').read()
view.addModel(pdb_str, 'pdb', {'hbondCutoff': hbondCutoff})
view.setStyle({"model": 0}, {'cartoon': {}})
view.setStyle({"model": 1}, {'cartoon': {'colorscheme': {'prop': 'b', 'gradient': 'roygb', 'min': 0, 'max': 100}}})
view.zoomTo()
view.show()
if num_designs > 1:
def on_change(change):
if change['name'] == 'value':
with output:
output.clear_output(wait=True)
plot_pdb(change['new'])
dropdown = widgets.Dropdown(
options=["best"] + [str(k) for k in range(num_designs)],
value="best",
description='design:',
)
dropdown.observe(on_change)
output = widgets.Output()
display(widgets.VBox([dropdown, output]))
with output:
plot_pdb(dropdown.value)
else:
plot_pdb()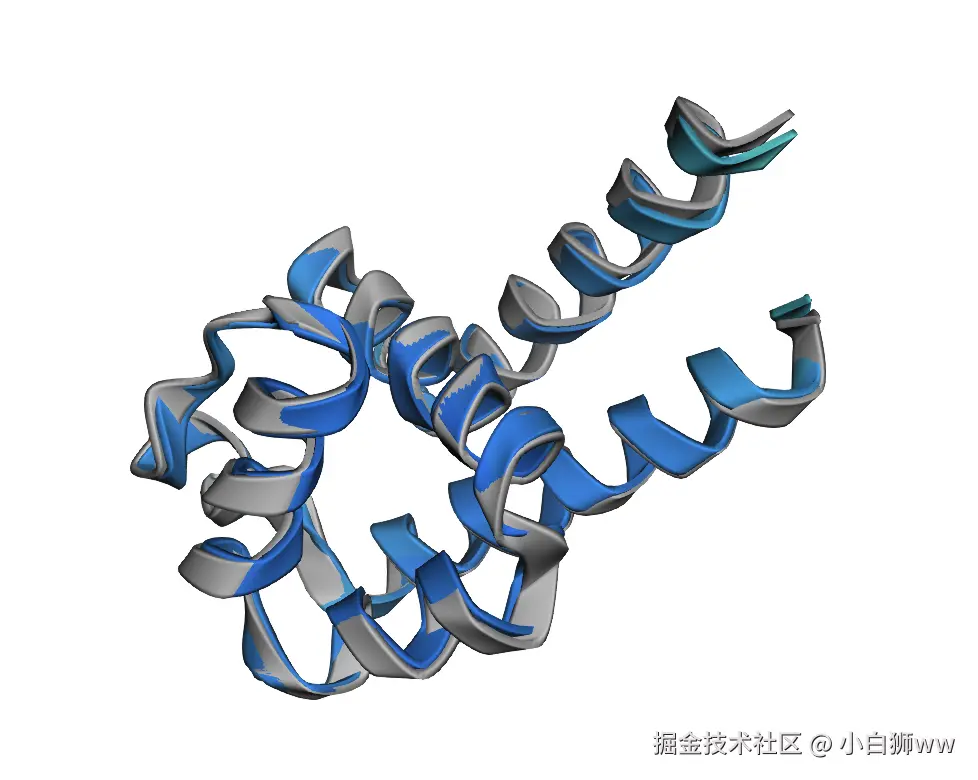
2.6 打包和下载
scss
def create_zip():
zip_path = f"{path}.result.zip"with zipfile.ZipFile(zip_path, 'w', zipfile.ZIP_DEFLATED) as zipf:
for root, _, files in os.walk("outputs"):
for file in files:
if file.startswith(path):
file_path = os.path.join(root, file)
arcname = os.path.relpath(file_path, "outputs")
zipf.write(file_path, arcname)
for root, _, files in os.walk("outputs/traj"):
for file in files:
if file.startswith(path):
file_path = os.path.join(root, file)
arcname = os.path.relpath(file_path, "outputs/traj")
zipf.write(file_path, arcname)
return zip_path
try:
zip_path = create_zip()
file_size = os.path.getsize(zip_path)/1024/1024
display(HTML(f'<b style="color:green">Compression completed!</b> File size: {file_size:.2f} MB'))
display(HTML(f'<b>Download link:</b> <a href="{zip_path}" download>{zip_path}</a>'))
except Exception as e:
display(HTML(f'<b style="color:red">An error occurred:</b> {str(e)}'))
display(HTML('Please check if the path exists: <code>!ls outputs/{path}*</code>'))Compression completed! File size: 0.50 MB
Download link: test_s67hh.result.zip