问题来源于读NicheCompass时,对cell-level 与 spot-level概念的迷惑Quantitative characterization of cell niches in spatially resolved omics data | Nature Genetics
- 图例说明(图 1a 及对应正文)
Birk et al. (2025)1 在图 1a 的图注中写道: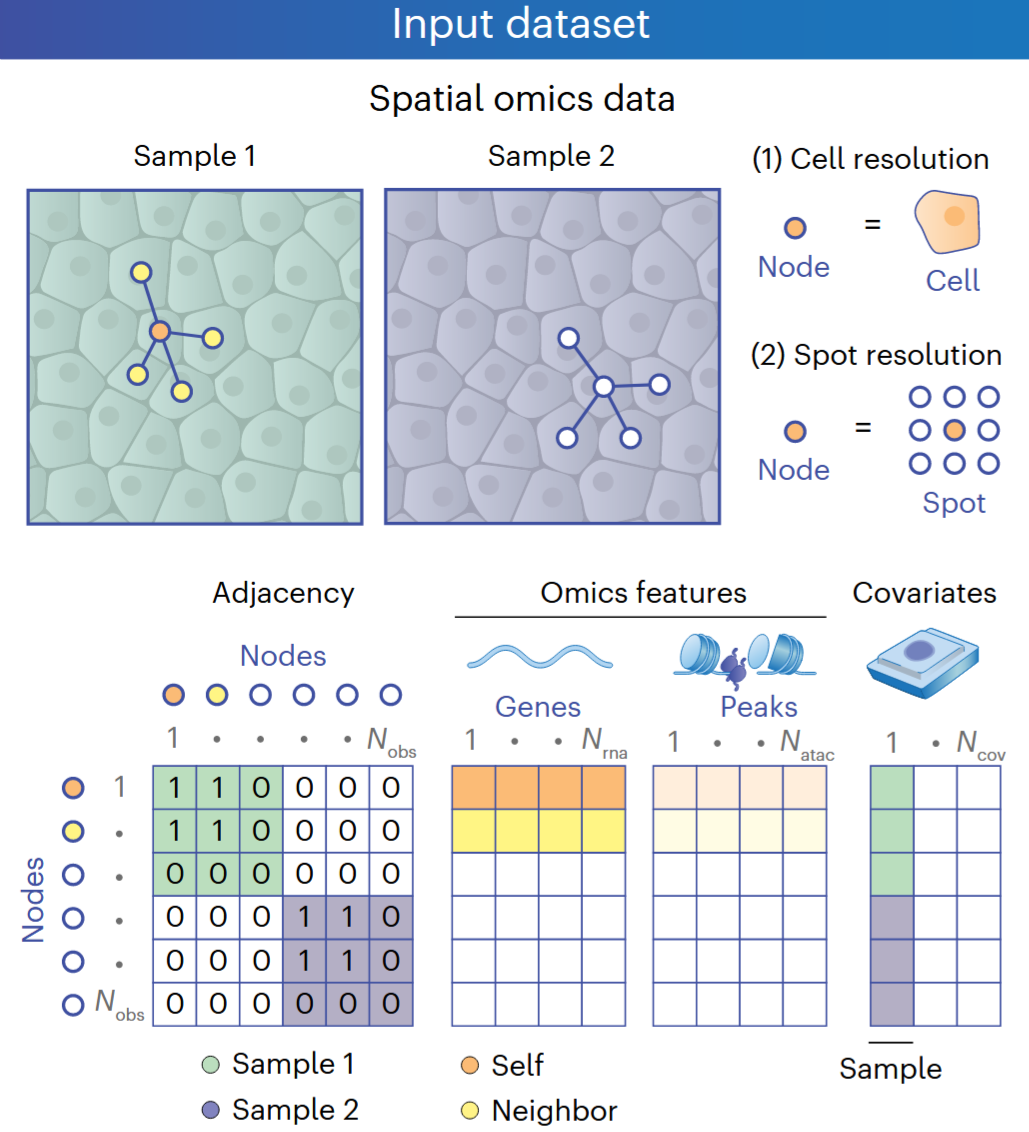
"NicheCompass takes single-sample or multi-sample spatial omics data with cell-level or spot-level observations as input... each cell or spot representing a node."
此处直接给出了两种输入粒度:单细胞(cell-level)与捕获点(spot-level)。
2.数据集描述部分(Methods 段:Data exclusions 与 Experiments)
在介绍所用数据时,作者列表指出:
"The Spatial ATAC--RNA seq mouse brain dataset included 9,215 spot-level observations ... The Stereo-seq mouse embryo dataset included 5,913 spot-level observations 。"
对比之下,其他如 seqFISH、STARmap PLUS 等则被称为"cell-level"或接近单细胞分辨率的数据。这进一步说明了"spot-level"与"cell-level"是两类不同分辨率的数据来源。
在空间组学(spatial omics)数据中,单细胞(cell-level)和 spot(spot-level)这两种观测尺度的主要区别体现在以下三方面,文献均在正文中给出了相应示例:
- 分辨率与数据精度
• 单细胞分辨率(cell-level)------每个观测节点对应一个完整细胞,因而能够直接捕获个体细胞的转录组甚至多组学特征,分辨精度为亚细胞级。
• Spot 分辨率(spot-level)------观测节点是"捕获区",其直径或像素大小通常在 10--100 µm(典型如 Visium、Stereo-seq,见作者原文 Methods 和数据描述方法部分),一个 spot 往往包含数个到数十个细胞的混合信号。 - 基因/特征数量与缺失率
• 单细胞数据集常可获得全转录组覆盖(作者所用 Slide-seqV2 记录约 4,000 基因方法数据描述),且表达矩阵稀疏但真实。
• Spot 数据为混合信号,基因总数通常更高(如 Spatial ATAC--RNA-seq 小鼠大脑使用 22,914 基因方法数据描述),但因整合多细胞,单个 spot 内基因的"真实来源"需要后期解卷积。 - 在 NicheCompass 建模中的处理策略
• 图构建:两种数据均可构建 k-近邻邻居图;对 spot 数据,作者指出"需进行 spot deconvolution"才能进一步提升结果(Discussion讨论中(4))。
• 模型表现:作者在补充图 14 及补充说明第 9 点(Supplementary Note 9)测试发现,NicheCompass 在 spot 尺度上的 niche coherence 和 spatial consistency 得分略低于同组织细胞级数据,原因在于混合信号稀释了真实信号。
综上,单细胞尺度提供"真值"级别细胞信息,适合精确研究 niche 边界和通信细节;spot 尺度提供组织上下文的大规模全景,但需额外解卷积或使用混合建模策略来弥补细胞异质性丢失。