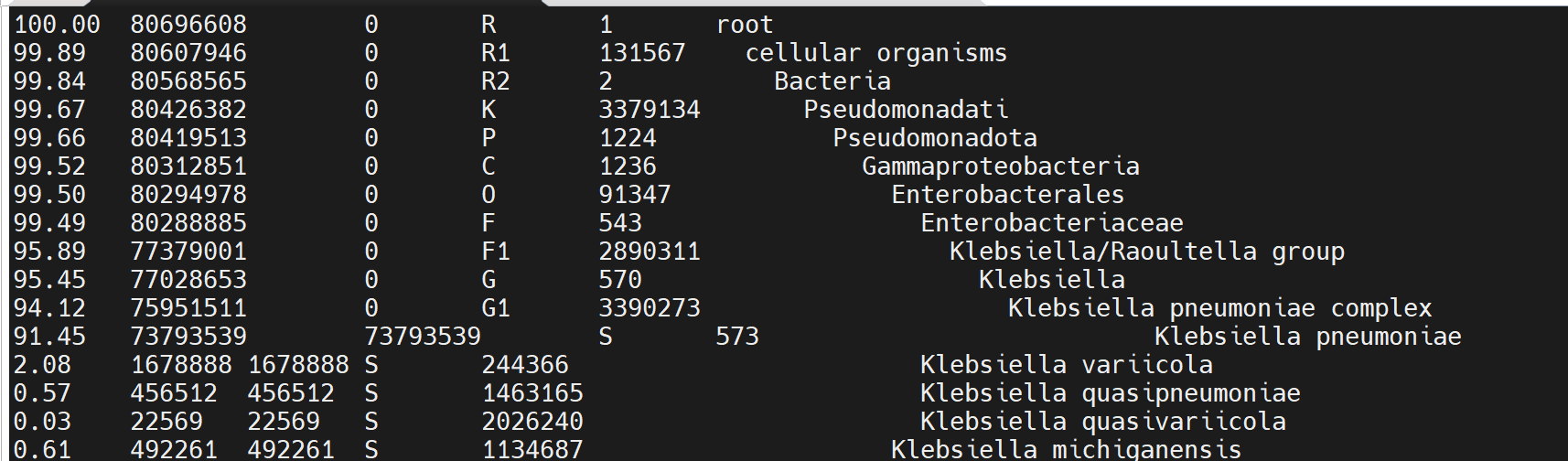
kraken2去除污染
conda create -n kraken2
conda activate kraken2
conda install kraken2 -c bioconda
mkdir kraken2_output
kraken2 --db ../../kraken2_db/k2_pluspf_20250402/ --threads 8 --paired 250811_HS67EV0804_R1.fastq.gz 250811_HS67EV0804_R2.fastq.gz --use-names --report-zero-counts --report ./kraken2_output/sample.report --output ./kraken2_output/sample.kraken
#######使用bracken查看species级别占比
bracken-build -d ../../../kraken2_db/k2_pluspf_20250402/ -t 8
srun bracken -d ../../../kraken2_db/k2_pluspf_20250402/ -i sample.report -o sample.bracken -r 150 -l S
less sample_bracken.report
##############使用KrakenTools提取目标reads############
conda install KrakenTools -y -c bioconda
srun extract_kraken_reads.py -k sample.kraken -s ../250811_HS67EV0804_R1.fastq.gz -s2 ../250811_HS67EV0804_R2.fastq.gz -o Kpn_573_R1.fastq -o2 Kpn_573_R2.fastq -t 573 --include-children --fastq-output --report sample.reportsample_bracken.report : S(species)--573为taxid
#####fastp#######
srun fastp \
-w 12 \ # 使用12个线程并行加速
-i ./raw_data/${sample}_1.fq.gz \ # 输入文件:R1
-I ./raw_data/${sample}_2.fq.gz \ # 输入文件:R2
-o ./clean_data/${sample}_1.fastp.fq.gz \ # 输出文件:清洗后的 R1
-O ./clean_data/${sample}_2.fastp.fq.gz \ # 输出文件:清洗后的 R2
-f 10 \ # 对 R1 每条 read,切除前10个碱基
-F 10 \ # 对 R2 每条 read,切除前10个碱基
-n 10 \ # 允许最多10个N,否则丢弃
-q 20 \
--dedup # 低于Q20的碱基视为低质量
--detect_adapter_for_pe \ # 自动检测并去除接头(推荐)
-h ./clean_data/${sample}.fastp.html \ # 输出 HTML 报告
-j ./clean_data/${sample}.fastp.json # 输出 JSON 报告(方便统计/批处理)
######fastqc质控#############参数----cov-cutoff可用于过滤contig##
srun fastqc HS67_1.clean.fq.gz HS67_2.clean.fq.gz -o clean_fastqc_report
#########spades拼接#############
srun spades.py -1 A18GX092_1.clean.fq.gz -2 A18GX092_2.clean.fq.gz --isolate -o spades_output
#############quast评估拼接质量#################
srun quast -t 16 -o quast_output spades_output/scaffolds.fasta
##质量评估标准:contig数目小于1000 , N50大于10bp 最小contig>200bp 评估污染:核苷酸ani平均一致性95% fastani 肺克GC含量为54-55%作业
#!/bin/bash
#SBATCH --job-name=spades_HS67 # 作业名称
#SBATCH --nodes=1 # 需要的节点数
#SBATCH --ntasks=1 # 总任务数
#SBATCH --cpus-per-task=20 # 每个任务的CPU核数
#SBATCH --mem=80G # 内存需求
#SBATCH --output=spades_%j.out # 标准输出日志
#SBATCH --error=spades_%j.err # 错误输出日志
运行SPAdes
spades.py -1 bac_HS67_1.clean.fq.gz -2 bac_HS67_2.clean.fq.gz --isolate -t 20 -o spades_out
概念解释
Kmer
k-mer 就是从 reads 中截取的长度为 k 的连续小片段。
例如 read 序列:ATGCGT,如果 k=3,就有 ATG、TGC、GCG、CGT 四个 3-mer。
组装器(如 SPAdes)用这些 k-mer 构建 de Bruijn 图,再拼接成 contigs。
k 值的大小 会直接影响组装的碎片化程度、错配率和连续性。
- 如果只用一个小 k(比如 21),装配可能非常连续,但错误也多;
- 如果只用一个大 k(比如 77),重复区能拆开,但低覆盖区会组装不出来。
SPAdes 默认的 -k 21,33,55,77 表示用不同长度的 k-mer 分阶段构建组装图:
小 k 保证敏感性,
大 k 提升连续性
N50
N50 = 一种衡量组装连续性的统计量
把所有 contig 从长到短排序;依次把它们的长度加起来,直到累计长度达到总组装长度的 50%;此时那个 contig 的长度,就是 N50。
举例:
假设组装结果有 5 条 contig,长度如下(单位:kb):
Contig1 = 200
Contig2 = 150
Contig3 = 80
Contig4 = 40
Contig5 = 30
总长度 = 500 kb
排序后累加:
Contig1 = 200 → 占 40%
Contig2 = 150 → 累计 350 → 占 70% ≥ 50%
所以 N50 = 150 kb。
解释:一半以上的基因组长度,来自于长度 ≥ 150 kb 的 contigs。
越大越好:N50 大说明组装越连续,contig 越长。
L50
达到总长度一半所需的 contig 数量
举例:
设 contig 长度(kb):200, 150, 80, 40, 30;总长 = 500 kb。
从大到小累加:
200(占 40%)→ 未到 50%
200+150=350(占 70%)→ 首次 ≥50%
此时累计用了 2 条 contig,所以 L50=2
越小越好
L50 越小越好,说明少数长 contig 就能覆盖一半基因组。
以下是一个细菌基因组拼装完成后的quast报告:
可以看到total length为5.56Mb,largest contig为0.459Mb,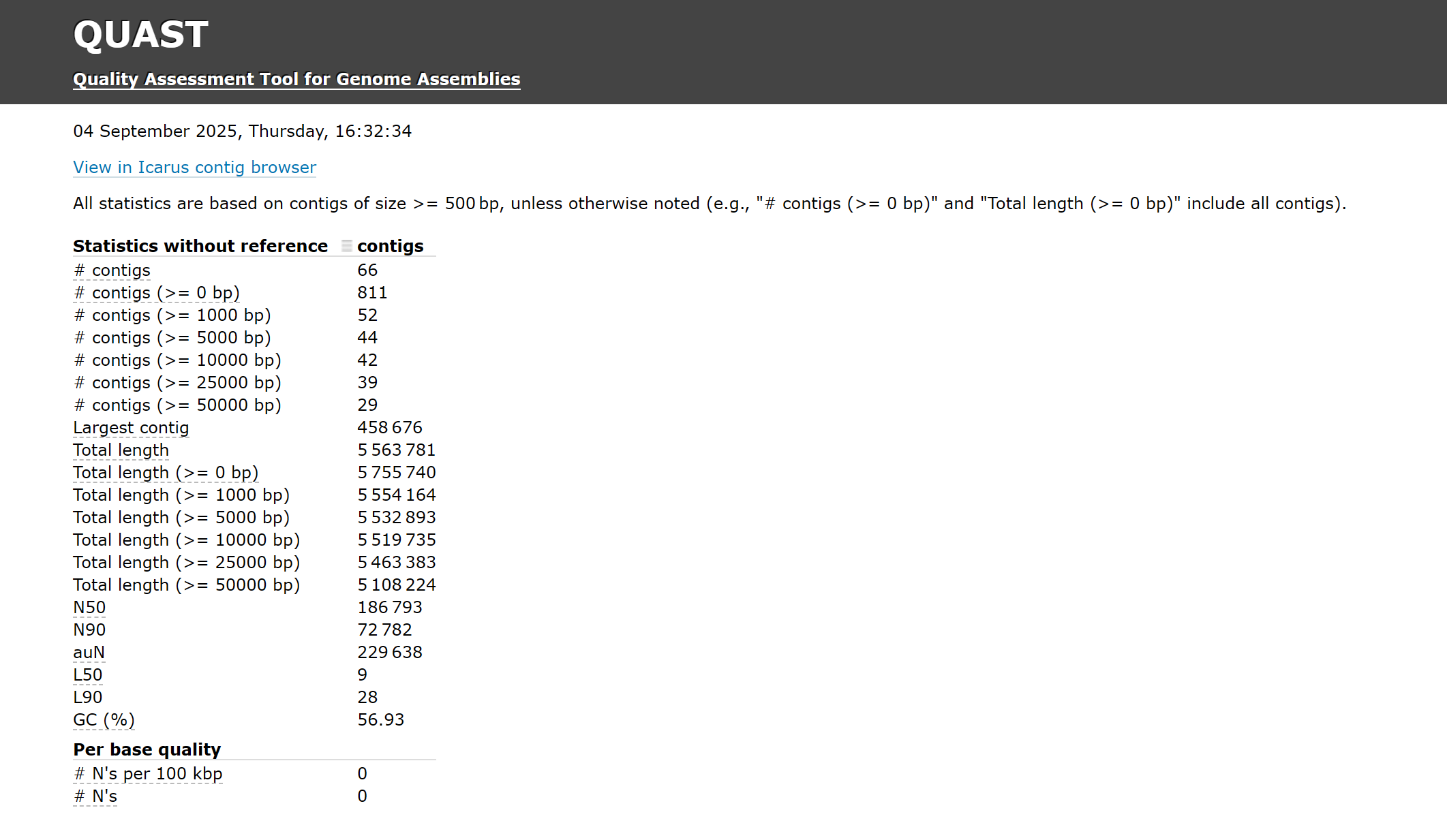
#############prokka注释############
prokka spades_output/scaffolds.fasta --outdir prokka_output --prefix A18GX092 --kingdom Bacteria --locustag A18GX092 --compliant --force注释所得结果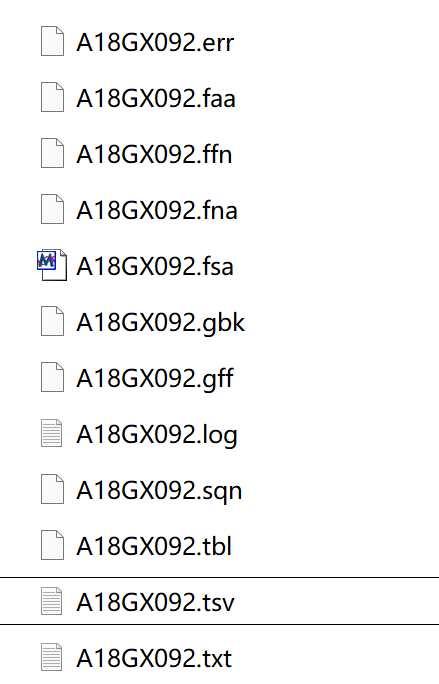
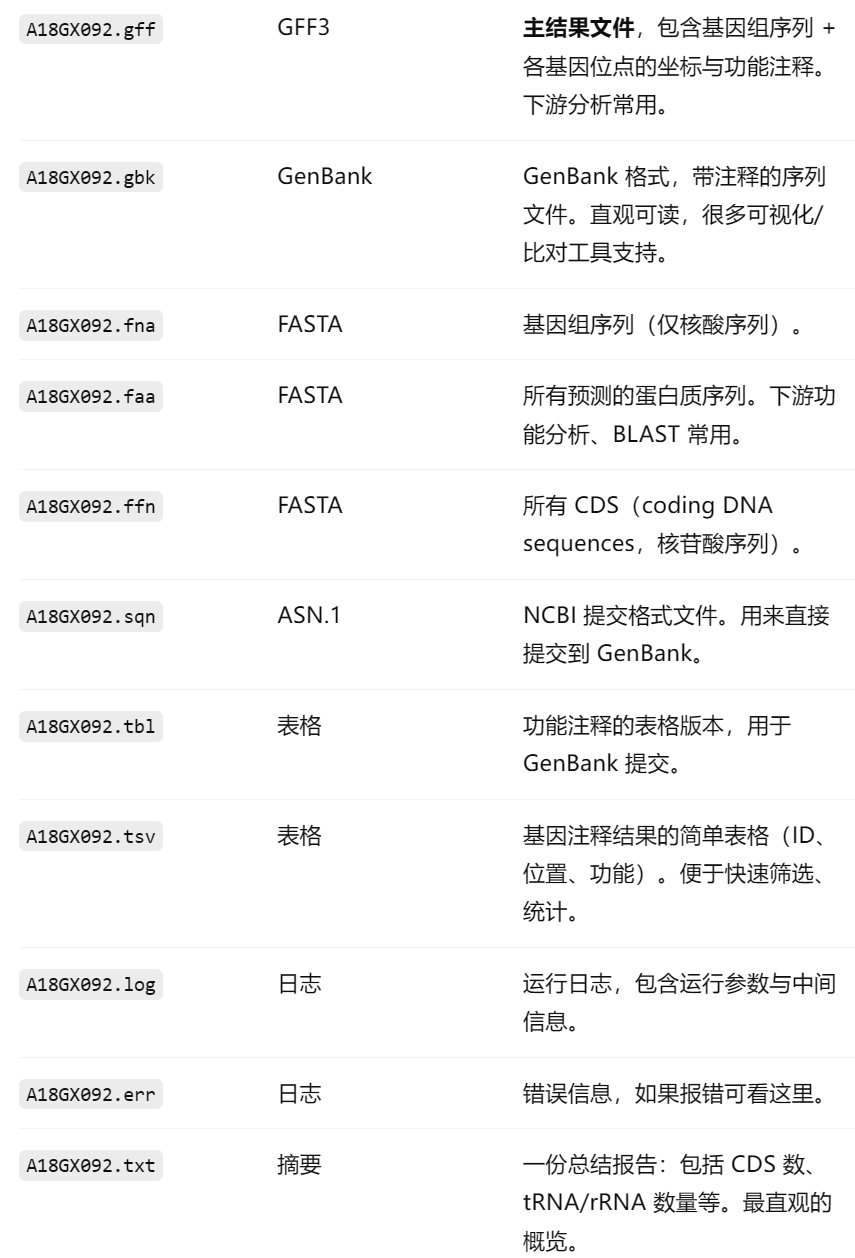
tsv文件包含内容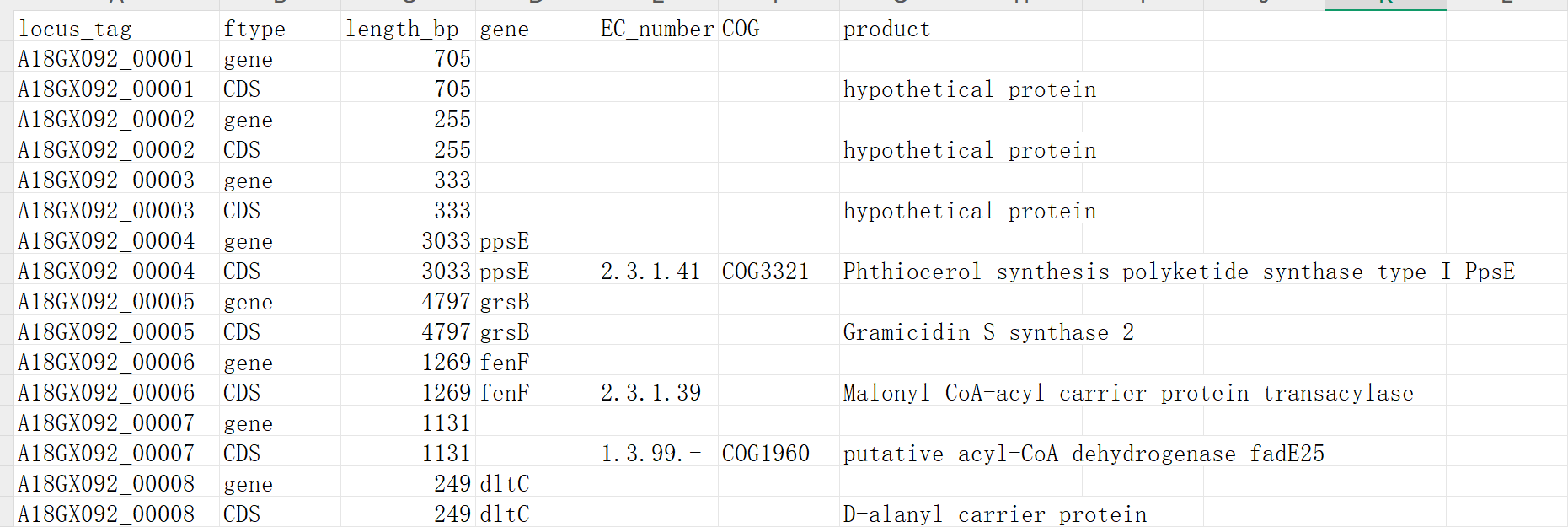
txt文件内容
gff文件内容
开头
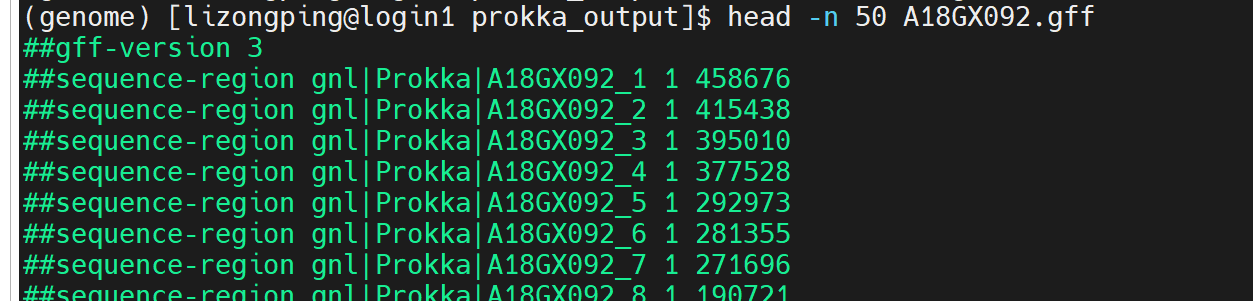
##gff-version 3→ 标准 GFF3 文件头。
##sequence-region→ 定义每条 contig 的名称和长度(比如A18GX092_1长度 458,676 bp)
中间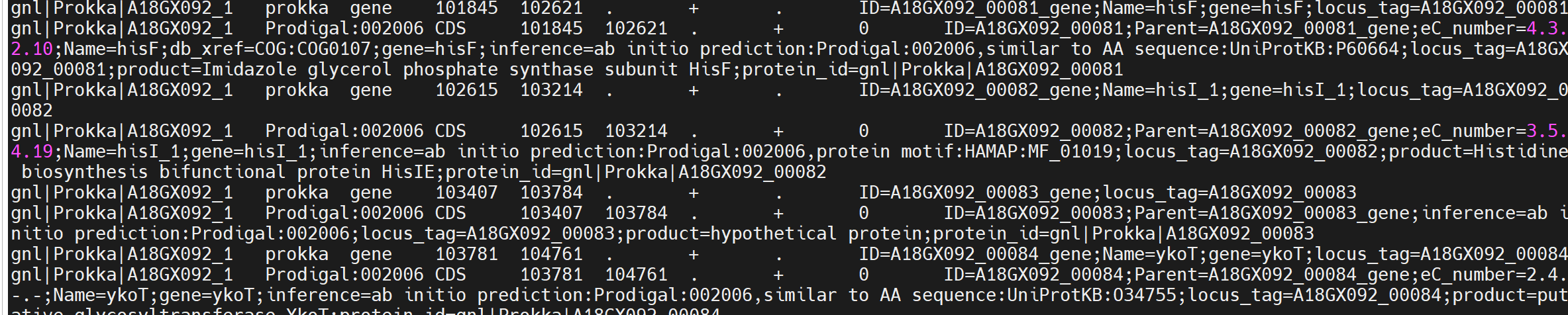
GFF 文件标准列(9 列)解释:
- seqid:所在的 contig 名称(如 A18GX092_1)。
- source:注释来源,这里是 Prokka。
- type:特征类型(CDS, gene, rRNA, tRNA, misc_feature 等)。
- start:起始坐标。
- end:终止坐标。
- score:得分(一般用不到,常为 .)。
- strand:所在链,+ 或 -。
- phase:阅读框,0/1/2(只对 CDS 有意义)。
如果 CDS 起点不同,phase 的值不同:
CDS 从 ATG 开始 → phase = 0 (对齐完整 codon)
CDS 从 ATG 开始 → phase = 2 (要跳过 2 个碱基,才能对齐完整 codon)
CDS 从 ATG 开始 → phase = 1 (要跳过 1 个碱基,才能对齐完整 codon)
- attributes:附加信息,如基因 ID、功能描述、EC/COG 等。
结尾

原始 contigs 的碱基序列,与 .fna、.fsa 内容相同