文章目录
介绍
最近,依据美国医学遗传学与基因组学学会(ACMG)和分子病理学协会(AMP)的指导原则,并遵循 ClinGen 关于变异分类的建议,开发出了一种全基因组变异效应校准方法。尽管全基因组方法具有临床应用价值,但新的证据表明,需要针对基因和特定环境进行校准以提高准确性。基于以往的工作,我们开发了一种算法,用于将来自变异效应多重检测(MAVE)和计算变异效应预测器(VEP)的功能评分转换为 ACMG/AMP 证据强度。
我们的方法旨在确保在不同基因和评分分布的情况下都能保持稳定的性能,所有变量均从输入数据中自适应地确定,从而避免了可能因人为调整或过度拟合而导致的证据强度超出实际支持范围的情况。为了便于应用,我们引入了 acmgscaler,这是一个轻量级的 R 软件包以及一个可即插即用的 Google Colab 笔记本,用于对自定义数据集进行校准。这一算法框架将 MAVEs/VEPs 与临床可操作的变异分类之间进行了连接。
A genome-wide variant effect calibration method was recently developed under the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP), following ClinGen recommendations for variant classification. While genome-wide approaches offer clinical utility, emerging evidence highlights the need for gene- and context-specific calibration to improve accuracy. Building on previous work, we have developed an algorithm tailored to converting functional scores from both multiplexed assays of variant effects (MAVEs) and computational variant effect predictors (VEPs) into ACMG/AMP evidence strengths.
Our method is designed to deliver consistent performance across different genes and score distributions, with all variables adaptively determined from the input data, preventing selective adjustments or overfitting that could inflate evidence strengths beyond empirical support. To facilitate adoption, we introduce acmgscaler, a lightweight R package and a plug-and-play Google Colab notebook for the calibration of custom datasets. This algorithmic framework bridges the gap between MAVEs/VEPs and clinically actionable variant classification.
为了建立统一的临床遗传和基因组检测方法,美国医学遗传学与基因组学学会以及分子病理学协会(ACMG/AMP)制定了评估常染色体疾病基因变异的致病性和良性性的指导原则(里奇斯等人,2015 年)。此后,这些指导原则在概率框架内得到了正式确立,展示了 ACMG/AMP 将临床证据进行组合的准则与致病性的可能性比值是如何相一致的(塔维蒂安等人,2018 年)。在这个框架内,每个分类(支持、中度、强烈和非常强烈)的证据强度已被定义为呈指数增长。该模型为系统地验证和校准致病性证据提供了基础,推动了对变异分类的改进,并将预测工具的作用从支持性证据扩展到更高级别的证据(布尼奇等人,2019 年)。
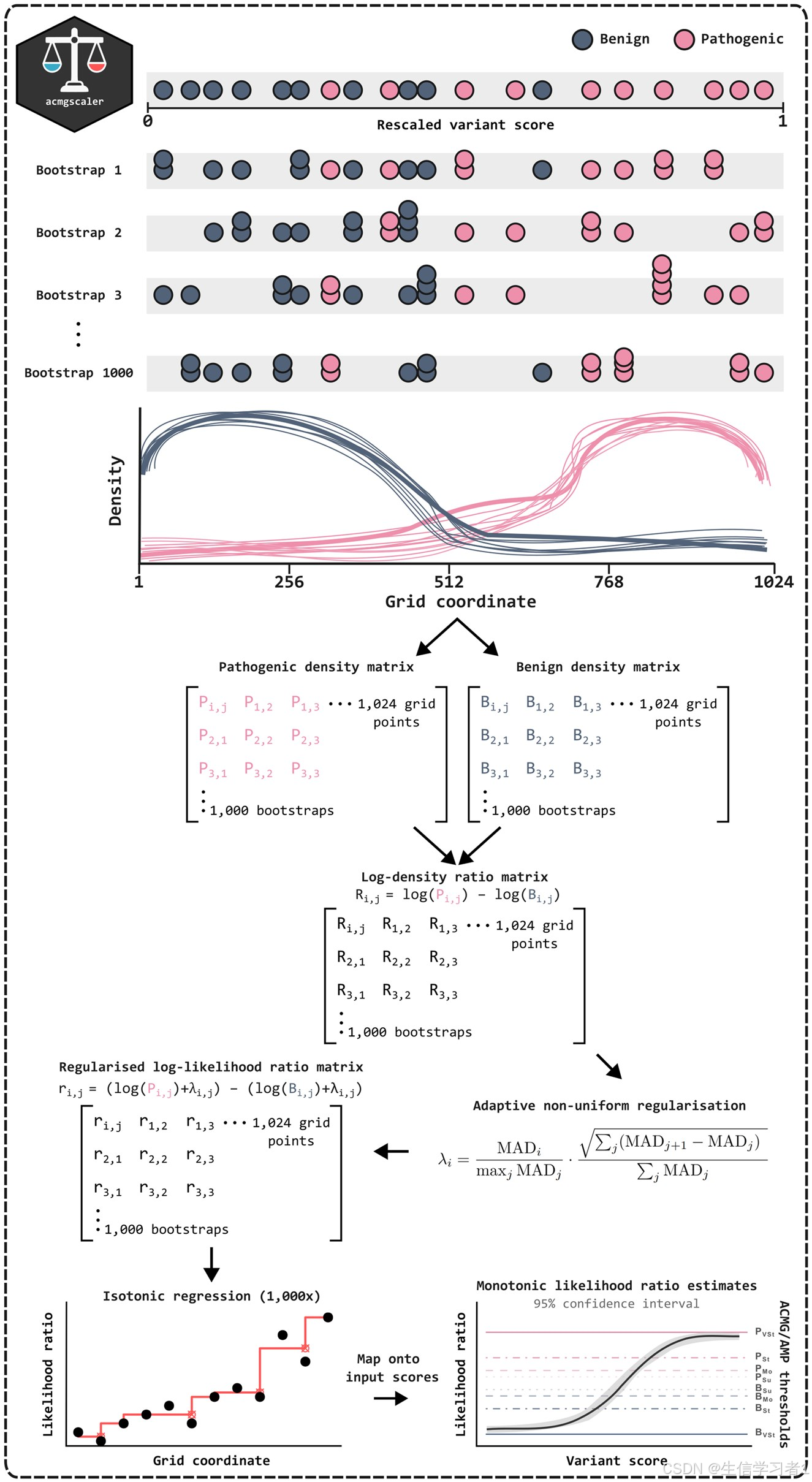
其中一种扩展适用于计算型变异效应预测器(VEP),其中对证据的适当加权对于确保准确分类至关重要。最近,佩贾沃等人开发了一种方法,通过将窄滑动窗口应用于已按致病性或良性进行标注的得分列表,来估算变异的致病性与良性性的密度比率(佩贾沃等人,2022 年)。由此得出的致病性几率,相当于阳性似然比(LR+;以下简称为 LR),可以与由 ACMG/AMP 框架在预先设定的致病性概率前提下确定的证据阈值进行比较(里查兹等人,2015 年;塔维蒂安等人,2018 年;布尼奇等人,2019 年)。使用这种方法,作者估计了针对 13 种广泛使用的 VEP 的每个证据水平的全基因组阈值,即得分区间。此后,这项工作又通过对其他 VEP 的校准而得到了扩展(伯格奎斯特等人,2025 年)。
将全基因组校准应用于变异分类的 VEP 分数已显示出显著的益处。例如,一项研究在 300 名疑似患有罕见疾病的患者中使用了全基因组阈值,发现与仅考虑已知的疾病相关基因的情况相比,达到具有强烈致病性证据的变异数量增加了 2.6 倍(Stenton 等人,2024 年)。重要的是,该分析得出结论,全基因组校准不太可能导致将过多的变异错误地分类为致病性或很可能致病性。然而,其他人对全基因组校准的局限性提出了担忧,特别是在掩盖不同基因和遗传模式下的表现方面(迪亚斯等人,2024 年,伊萨科夫等人,2024 年,特朱拉等人,2024 年)。这些发现表明,仅依靠全基因组校准可能不足以解决这些差异,并强调需要一种针对基因和特定情况的校准方法。
变异效应多重检测(MAVE)能够提供一种无偏的功能性证据来源(Weile 和 Roth 2018、Fowler 等人 2023、Rubin 等人 2025),为改进胚系基因组变异的分类(特别是那些经常被归类为意义不确定的罕见错义变异)提供了巨大潜力(Chen 等人 2023)。然而,与 VEP 的输出类似,MAVE 的原始分数在不同基因中的范围和分布各不相同,并且无法直接对应于 ACMG/AMP 的证据强度。
代码
https://github.com/badonyi/acmgscaler
参考
- acmgscaler: an R package and Colab for standardized gene-level variant effect score calibration within the ACMG/AMP framework
- https://github.com/badonyi/acmgscaler