Pfam 数据库详解
一、Pfam 数据库核心介绍
(注:基础版本信息:Pfam 32.0,发布于 2018 年 9 月,涵盖 17929 个蛋白质结构域家族)
Pfam 38.0 (25,545 entries, 796 clans)
蛋白质的功能多样性往往源于其结构的复杂性,而结构域(domain)作为蛋白质中具备独立功能的核心区域,是解析蛋白质功能的关键切入点 ------ 不同结构域的组合与协同作用,造就了蛋白质丰富的生理功能。Pfam 数据库作为国际权威的蛋白质结构域家族数据库,其核心价值在于通过多序列比对和隐马尔可夫模型(HMM)预测,系统性整合并标注具有代表性的蛋白质结构域家族。
其中,Pfam 家族的核心条目为 Pfam - A,每个条目均包含三大核心组件:一是精选的种子序列比对集合,涵盖该家族少量具有代表性的成员序列,保障家族特征的精准性;二是基于种子比对构建的轮廓隐马尔可夫模型(profile HMMs),为序列识别提供算法基础;三是自动生成的全序列比对结果,囊括数据库中可检测到的该家族所有相关蛋白质序列,其范围由轮廓 HMM 搜索规则严格界定。
每个 Pfam 条目均围绕 6 类核心信息构建:家族(Family)、结构域(Domain)、重复序列(Repeat)、基序(Motifs)、卷曲螺旋(Coiled - Coil)及无序区域(Disordered)。为便于关联分析,具有序列、结构或轮廓 HMM 相似性的 Pfam 条目会被进一步归类为宗族(clan)。
近年来,Pfam 数据库借助机器学习技术实现了突破性拓展。2022 年,谷歌研究院研发的 ProtENN 模型为其新增约 680 万条蛋白质功能注释数据,这一数量相当于过去十年注释总量的总和,直接推动数据库注释的蛋白质序列占比提升近 10%,还成功预测了 360 种人类蛋白质的功能。在家族拓展方面,2023 年有研究通过实验验证,为 Pfam 数据库新增多个蛋白家族,并证实其中一个家族属于全新的翻译靶向毒素 - 抗毒素系统(TumE - TumA)超家族。此外,黄山学院柏晓辉博士发现并鉴定的 WW - like domain 全新结构域家族,也被 Pfam 收录并采纳为该家族的标准命名,凸显了数据库在整合前沿科研成果方面的时效性。
二、Pfam 数据库网页端使用指南
(一)"Jump to" 快速检索
该功能支持一键直达 Pfam 网站的目标条目,涵盖 Pfam 家族、宗族(clan)及 Uniprot 序列条目等核心内容,具备多类型 ID 识别能力,能帮助使用者跳过层级导航,快速定位所需信息。
(二)关键词精准搜索
Pfam 网站各主页顶部均设有搜索框,可通过关键词匹配目标 Pfam - A 家族。搜索范围全面覆盖数据库多维度信息,包括家族功能描述、Uniprot 序列条目及物种来源、PDB 数据库相关条目标题与主题、基因本体(GO)编号及条目,以及 InterPro 条目摘要等。为避免信息冗余,每个 Pfam - A 条目在搜索结果中仅显示一次,即便其在数据库多个分类下均有匹配记录。
(三)蛋白质序列定向搜索
通过该方式可借助 Pfam 的 HMMs 文库解析目标蛋白质的结构域构成。若待搜索序列存在于 Uniprot、NCBI Genpept 或 Pfam 已发布的微生物基因组序列集合中,可直接通过对应 ID 号检索;若为未收录序列,可选择两种检索方式:一是单序列搜索(Single protein search),直接粘贴序列至指定区域;二是批量搜索(Batch search),通过上传序列文件完成批量查询。
(四)蛋白质组专项分析
Pfam 已预先完成 Uniprot 蛋白质组中所有成员的结构域组成与结构计算。使用者可通过网站顶部的 "浏览" 链接,点击 "蛋白质组" 板块的字母索引获取完整蛋白质组列表。点击具体生物体名称,即可进入专属页面查看其蛋白质组的结构域组织形式与组成特征。此外,该模块的分类查询功能可助力快速筛选物种特异性结构域家族,不过此类跨物种比对操作耗时相对较长,需合理规划检索时间。
(五)特定结构域组合筛选
借助 Pfam 内置的结构域搜索工具,可精准筛选含特定结构域组合的蛋白质(如同时含 CBS 结构域与 IMPDH 结构域的蛋白质)。若需深入分析,可使用 PfamAlyzer 工具,该工具不仅能锁定目标结构域组合的蛋白质,还支持指定物种范围,并可设置结构域间的进化距离阈值,满足精细化研究需求。
三、本地 Pfam 搜索方案
当待分析的蛋白质序列数量庞大,或需保护序列隐私避免网络传输时,可通过pfam_scan.pl脚本实现本地搜索。执行该方案需提前配置三类核心资源:一是 HMMER3 软件,作为 HMM 模型运行的核心工具;二是 Pfam 的 HMM 文库及配套数据文件,保障序列比对的数据源;三是必要的 CPAN(综合 Perl 归档网络)模块,例如 Moose 模块等,确保脚本正常执行。
四、Pfam 常见问题解答
-
**家族划分至宗族(clan)的标准是什么?**划分采用结构化的多维度判定方式。若目标家族有已知结构,以结构特征为核心划分依据;若无结构信息,则通过三项标准综合判定:一是轮廓比对(如借助 HHsearch 工具);二是序列同一区域能否显著匹配两个不同 HMM;三是通过 SCOOP 方法挖掘搜索结果中体现家族关联的共性匹配。最终结合文献调研验证,如功能关联性等,确定家族的宗族归属。
-
Pfam_ls 与 Pfam_fs 文件的作用是什么? 二者是 Pfam 早期版本中配套的两类轮廓 HMM 模型。Pfam_ls 对应局部匹配模式,允许序列仅匹配 HMM 的部分区域;Pfam_fs 为全局匹配模式,要求序列与 HMM 全长完全匹配。在 HMMER2 版本中,二者联合使用可提升搜索灵敏度,但 HMMER3 版本的局部模式经优化后,已能达到同等灵敏度。因此,当前数据库不再提供这两类专用文库,仅保留统一的
Pfam - A.hmm蛋白质文库。 -
**iPfam 的核心功能是什么?**iPfam 聚焦于存储 PDB 数据库中蛋白质结构域的互作信息。对于含多个结构域的蛋白质结构,该数据库会先判断各结构域间距离是否满足互作条件,若符合,则进一步计算形成互作的化学键特征,为蛋白质互作机制研究提供基础数据。
-
**全局比对中 "-" 和 "." 的区别是什么?**两者均用于表示序列比对中的空位,但含义不同。"-" 代表 HMM 比对中以缺失状态替代匹配状态,表明序列该位置缺少 HMM 模型预期的氨基酸残基;"." 用于填补因 HMM 插入状态导致的序列空位。在 HMM 中插入状态以 "I" 标记,对应序列插入位置的残基统一用小写字母表示。
-
**比对结果中 SS 行代表什么?**SS 行用于呈现蛋白质的二级结构信息,其数据源自 DSSP 程序的分析结果。DSSP 程序由 Wolfgang Kabsch 和 Chris Sander 开发,专门用于为 PDB 数据库中蛋白质分配二级结构,仅负责结构解析而非预测。SS 行中各类符号含义如下:C 代表随机卷曲、H 代表 α 螺旋、G 代表 3 (10) 螺旋、I 代表 π 螺旋、E 代表氢键 β 链(延伸链)、B 代表分离 β 桥中的残基、T 代表氢键合匝(3 匝、4 匝或 5 匝)、S 代表弯曲(以残基 i 为中心的五残基弯曲)。
五、相关数据库补充
Pfam 常与多个蛋白质相关数据库联动,共同构成完整的蛋白质结构与功能分析体系,核心关联数据库如下:
| 数据库名称 | 核心特征 |
|---|---|
| PROSITE | 基于序列相似性划分蛋白质家族,聚焦进化保守区域,含千余种家族和结构域的模式与轮廓信息,可通过蛋白质标记推测新测序蛋白质功能 |
| PRINTS | 以蛋白质指纹(fingerprints)为核心,由一组非重叠且序列上分离的保守基序组成,相比单一基序,能更灵活地编码蛋白质折叠方式与功能特征 |
| SMART | 涵盖 500 余个信号、胞外及染色质相关结构域家族,注释信息全面。分两种模式:常规模式适配 Swiss - Prot 等数据库,基因组模式适配完整测序基因组 |
| ADDA | 通过算法自动完成蛋白质结构域拆分与结构家族聚类,提升结构域分类的效率 |
| InterPro | 整合 Pfam、Prints、SMART 等多个数据库信息,实现蛋白质相关数据的一站式查询 |
| CDD | 即保守结构域数据库,核心数据源自 Pfam 与 SMART,专注于保守结构域的整合与检索 |
六、Pfam 核心术语释义
- Alignment coordinates(比对坐标):HMMER3 会输出两组结构域匹配坐标。外包络坐标(envelope coordinates)通过概率确定序列上的匹配区间;比对坐标则精准界定序列与轮廓 HMM 的最优比对区域。
- Architecture(结构):指单个蛋白质中所有结构域的组合形式与排列特征。
- Clan(宗族):由具有序列、结构或轮廓 HMM 相似性的 Pfam 条目组成的集合。
- Domain(结构域):蛋白质中具备独立结构与功能的基本单元。
- Domain score(结构域得分):单个结构域与 HMM 模型比对的量化结果。需注意,HMMER2 中多结构域序列得分为各结构域得分总和,此规则不适用于 HMMER3。
- DUF:即未知功能的结构域,特指已鉴定但功能尚未明确的蛋白质结构区域。
- Full alignment(完全比对):指得分高于 Pfam 对应条目 HMM 模型手动设定阈值的相关序列集合。
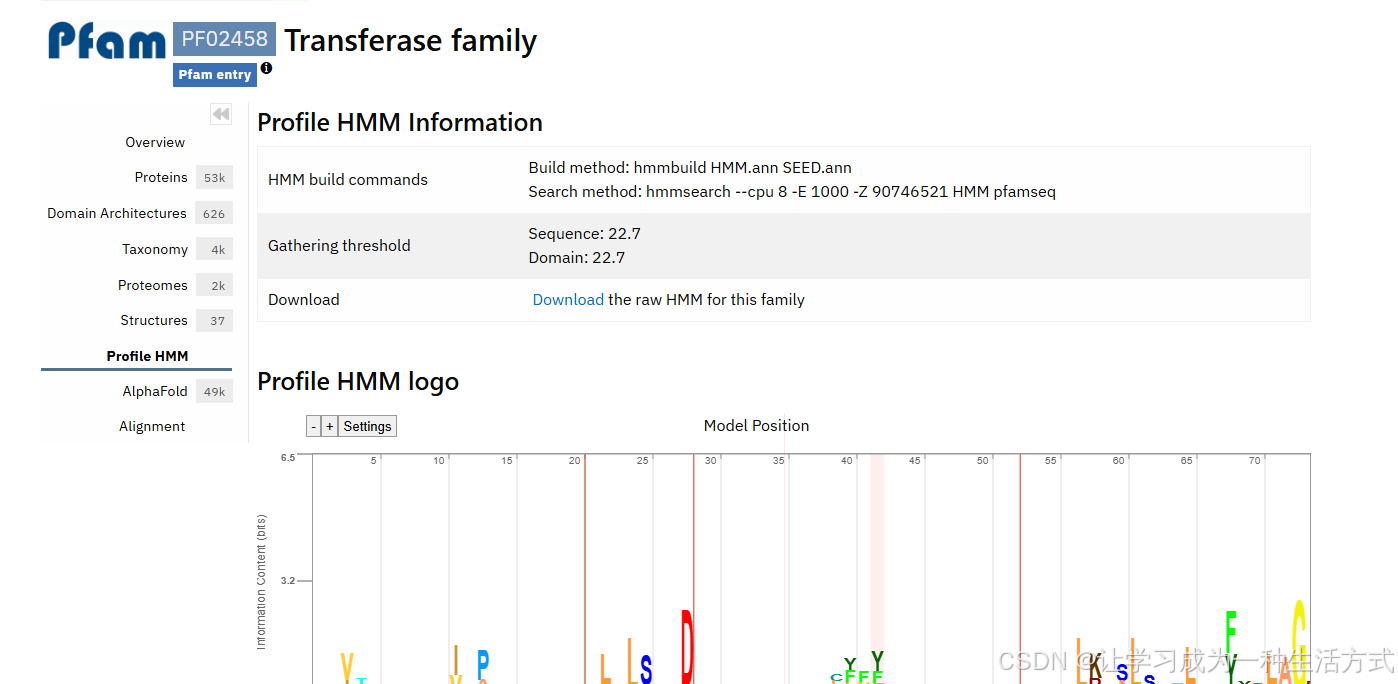
- Gathering threshold(GA,聚类阈值):构建完全比对的核心阈值,由家族构建者设定,序列需满足该阈值才可纳入完全比对范围,每个 Pfam HMM 均包含序列和结构域两类聚类阈值。
- HMMER:Pfam 用于构建和搜索 HMM 模型的核心程序。
- Metaseq:汇集各类宏基因组数据集的序列资源库。
- Noise cutoff(NC,噪声阈值):未纳入完全比对的序列中的最高匹配得分。
- Pfam - A:基于少量代表性序列手工注释构建的 HMM 条目,每个模型均手动设定阈值,并用于 Uniprot 数据库的序列检索。
- Posterior probability(后验概率):HMMER3 为轮廓 HMM 中插入和匹配状态的每个残基生成的可信度指标,值越高比对结果越可靠。可信度以 10(用 "*" 标记)至 1 的梯度表示,数据库中通过热图可视化,绿色代表高可信度,红色代表低可信度。
- Seed alignment(种子比对):Pfam 条目中代表性序列的比对集合,是构建该条目 HMM 模型的基础。
- Trusted cutoff(TC,可信阈值):完全比对中的最低匹配得分。
- E - values 和 Bit - scores:均为序列比对的量化指标。E 值代表随机序列获得同等或更高得分的概率,优质结果的 E 值通常远小于 1,且受数据库规模影响;比特值(Bit - scores)不受数据库规模干扰,Pfam 为每个家族设定比特值聚类阈值,达标序列可纳入完全比对。其中比特值 20 约对应 E 值 0.1,比特值 25 约对应 E 值 0.01 。
参考文献
Pfam: The protein families database in 2021: J. Mistry, S. Chuguransky, L. Williams, M. Qureshi, G.A. Salazar, E.L.L. Sonnhammer, S.C.E. Tosatto, L. Paladin, S. Raj, L.J. Richardson, R.D. Finn, A. Bateman, Nucleic Acids Research (2021) doi: 10.1093/nar/gkaa913