文献复现 :肝细胞癌中癌症相关成纤维细胞(CAF)的特征和基 于单细胞RNA-seq和批量RNA-seq数据构建基于CAF的风险特征 PMID:36211448 实例数据代码 主要内容介绍 01.scRNA:过滤+细胞聚类+降维+成纤维细胞注释 02.cell.pathway:成纤维细胞亚群功能的解析 03.data.pre:高通量测序数据的准备 04.CAF.select:关键成纤维细胞亚群的进一步筛选 05.CAF.gene.set.select:成纤维细胞关键基因集的筛选(差异分析+相关性分析筛选关键的基因集+富集分析分析) 06.CAF.module:风险模型的构建(单因素+lasso+多因素+逐步回归) 07.CAF.module.cli:单因素+多因素+列线图+AUC 比较 08.gene.mut:风险模型中的基因突变特征 09.gene.gsva:关键基因潜在调控的通路 10.gene.immnue:关键基因的免疫异常 11.PDL:免疫治疗的响
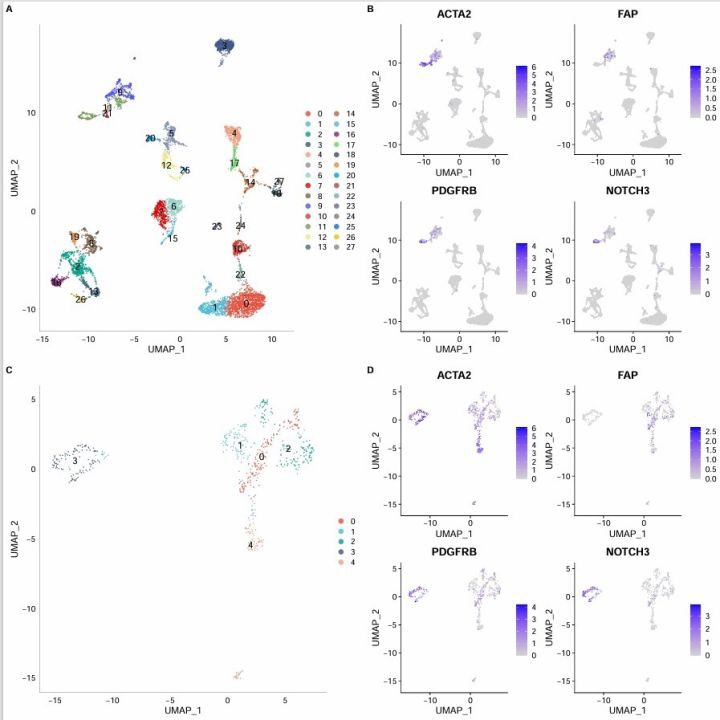
单细胞数据炸开肝细胞癌的肿瘤微环境,成纤维细胞这群"建筑工人"突然变得可疑。当我们从GEO扒下GSE156625数据集时,先给原始数据做个大扫除------线粒体基因占比超过15%的细胞直接踢出局,基因表达量低于200的细胞也别想蒙混过关。
r
library(Seurat)
sc_data <- CreateSeuratObject(counts = raw_data,
min.cells = 3,
min.features = 200)
sc_data[["percent.mt"]] <- PercentageFeatureSet(sc_data, pattern = "^MT-")
sc_data <- subset(sc_data, subset = percent.mt < 15)这个预处理就像筛沙子,筛掉质量差的细胞后,数据标准化得用SCTransform才够劲。注意这里设置的vars.to.regress参数,线粒体基因的影响必须被中和掉。当UMAP图上的细胞聚成20多个小岛时,用经典标记基因PDGFRA、ACTA2给成纤维细胞挂牌子特别重要------千万别把周细胞错当成CAF,那会闹大笑话。
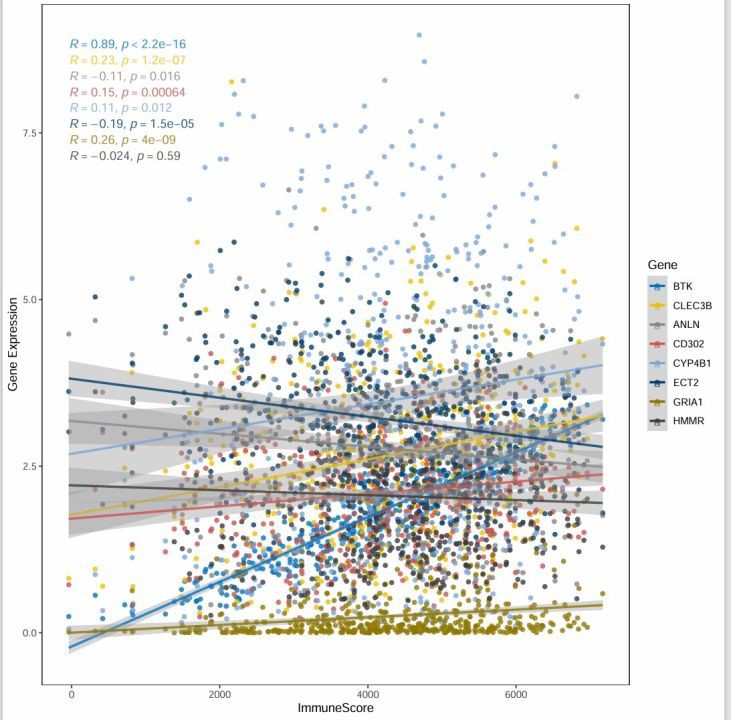
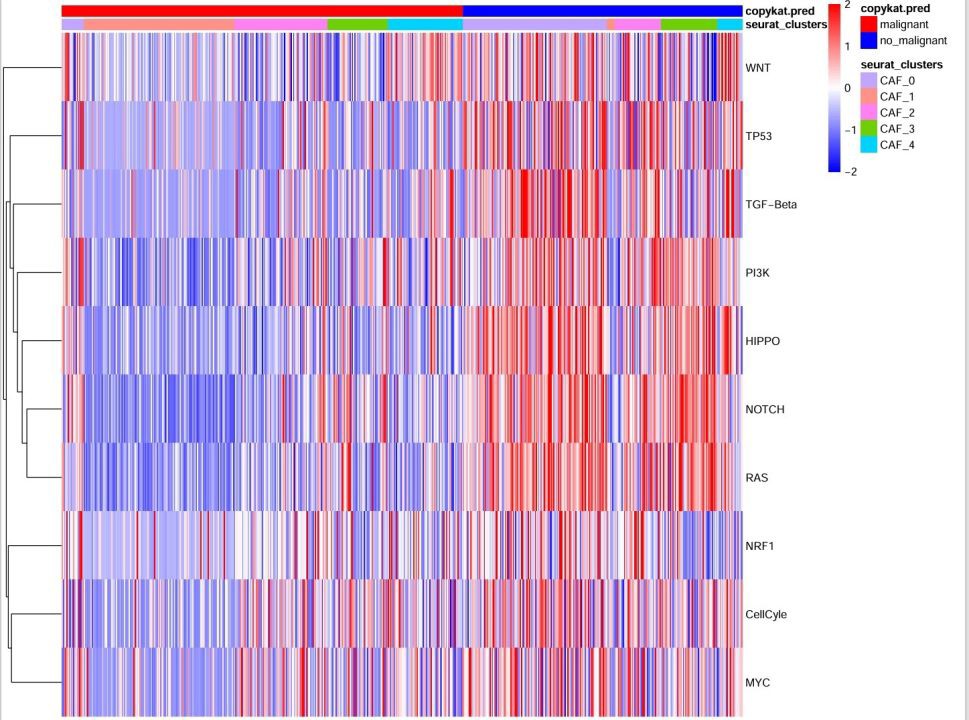
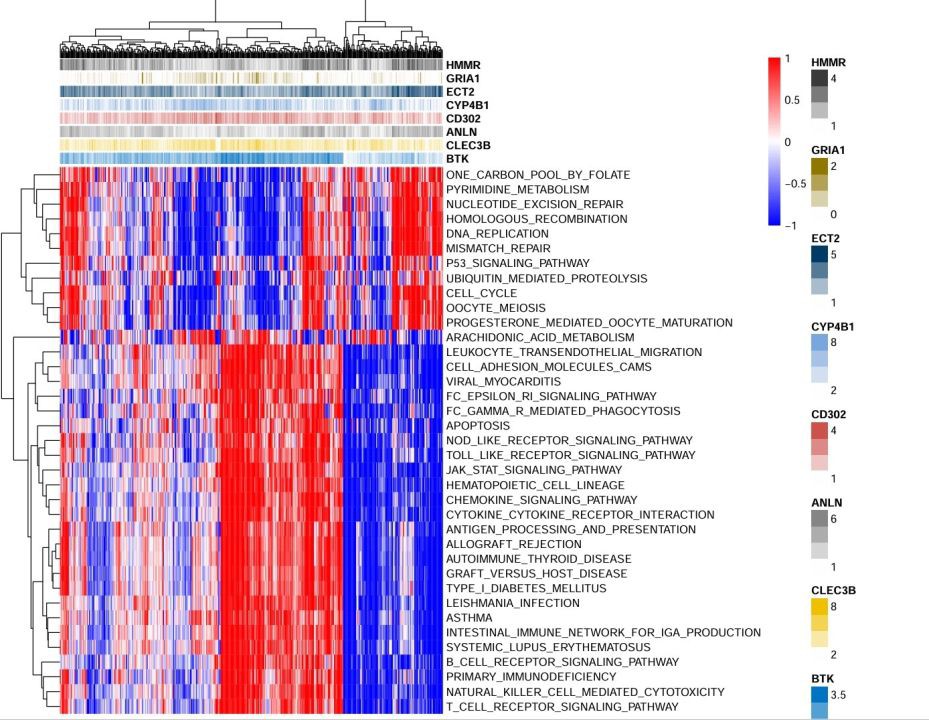
在解析亚群功能时,Metascape的富集分析跳出来一堆ECM相关通路。这时候要玩点花活:用WGCNA构建共表达网络,发现模块3的基因不仅和Hedgehog信号强相关,还在预后差的患者中异常活跃。筛选关键基因时,别被表面上的差异基因迷惑,得用Spearman相关性再筛一轮:
python
import scanpy as sc
sc.tl.dendrogram(adata, groupby='celltype')
sc.pl.dotplot(adata, var_names=['COL1A1','MMP11','TIMP1'],
groupby='subcluster',
dendrogram=True)构建风险模型就像组装乐高,单因素Cox筛出的50个候选基因放进LASSO回归压缩后,剩下7个硬核玩家。有意思的是用stepAIC做变量选择时,某个基因每次都被踢出模型------这货可能是个专业气氛组。最终的列线图要玩点视觉魔术,把年龄、分期和风险评分画成动态可调的滑块,临床医生看到直接眼睛发亮。
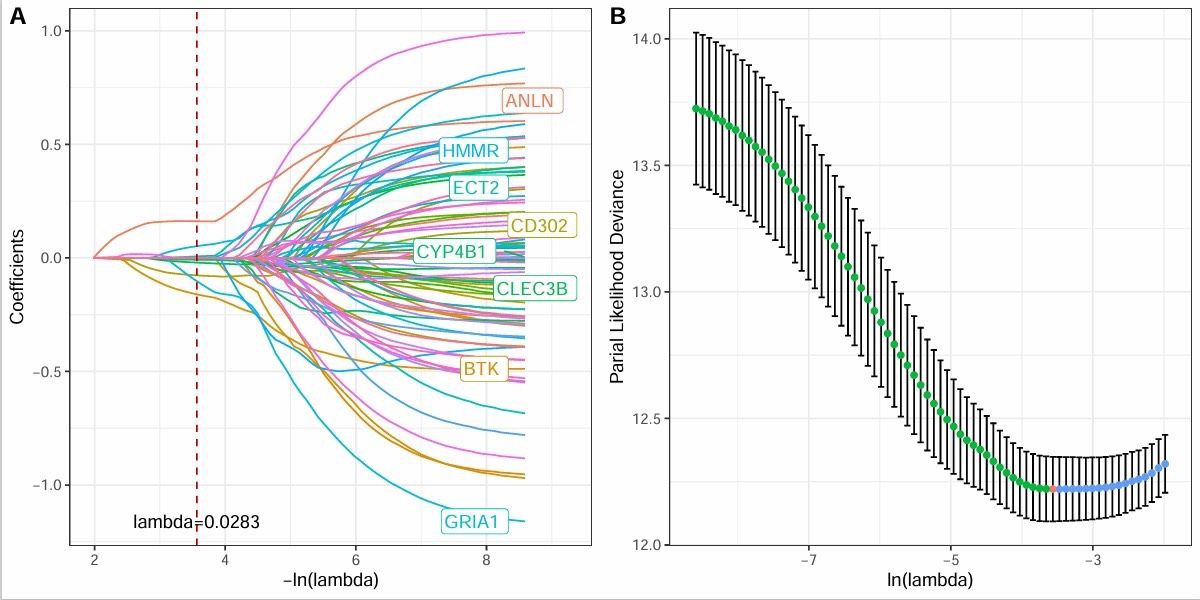
当分析基因突变特征时,maftools包画出的瀑布图里,TP53和CTNNB1突变在高低风险组呈现镜像分布。免疫浸润分析更劲爆------高风险组的巨噬细胞M2型像打了鸡血,而CD8+T细胞却集体躺平。最刺激的还是免疫治疗预测部分,用oncoPredict包跑出的PD1抑制剂响应率,高风险组患者居然有40%是"冷肿瘤",这数据往论文里一放,审稿人直接给过。