作者,Evil Genius
今天在县城遇到了初中的同桌,女同学,不知不觉快20年过去了。
多组学已经在路上,除了之前一直强调的基因组 + 单细胞空间 + 分子对接/分子动力学之外,代谢组学也慢慢提上了日程。
今天我们分享顶刊,国产顶刊

知识积累
调节性T细胞以表达FOXP3为特征,在抑制抗肿瘤免疫反应中起着关键作用。随着肿瘤进展,调节性T细胞在肿瘤微环境中逐渐积累,而效应T细胞数量却呈反向下降。
高度活跃的细胞能量代谢是癌症的重要特征之一,其通过驱动ATP生成来支持疾病发展。谷氨酰胺作为肿瘤细胞的主要能量来源,通过谷氨酰胺分解产生α-酮戊二酸,后者进入三羧酸循环为ATP生成和蛋白质合成供能。除α-酮戊二酸外,谷氨酰胺分解的副产物氨也在肿瘤微环境中不断累积,对肿瘤内细胞产生毒性效应并调节其活性。
氨会诱导肿瘤微环境中CD8+效应T细胞死亡或进入耗竭状态。
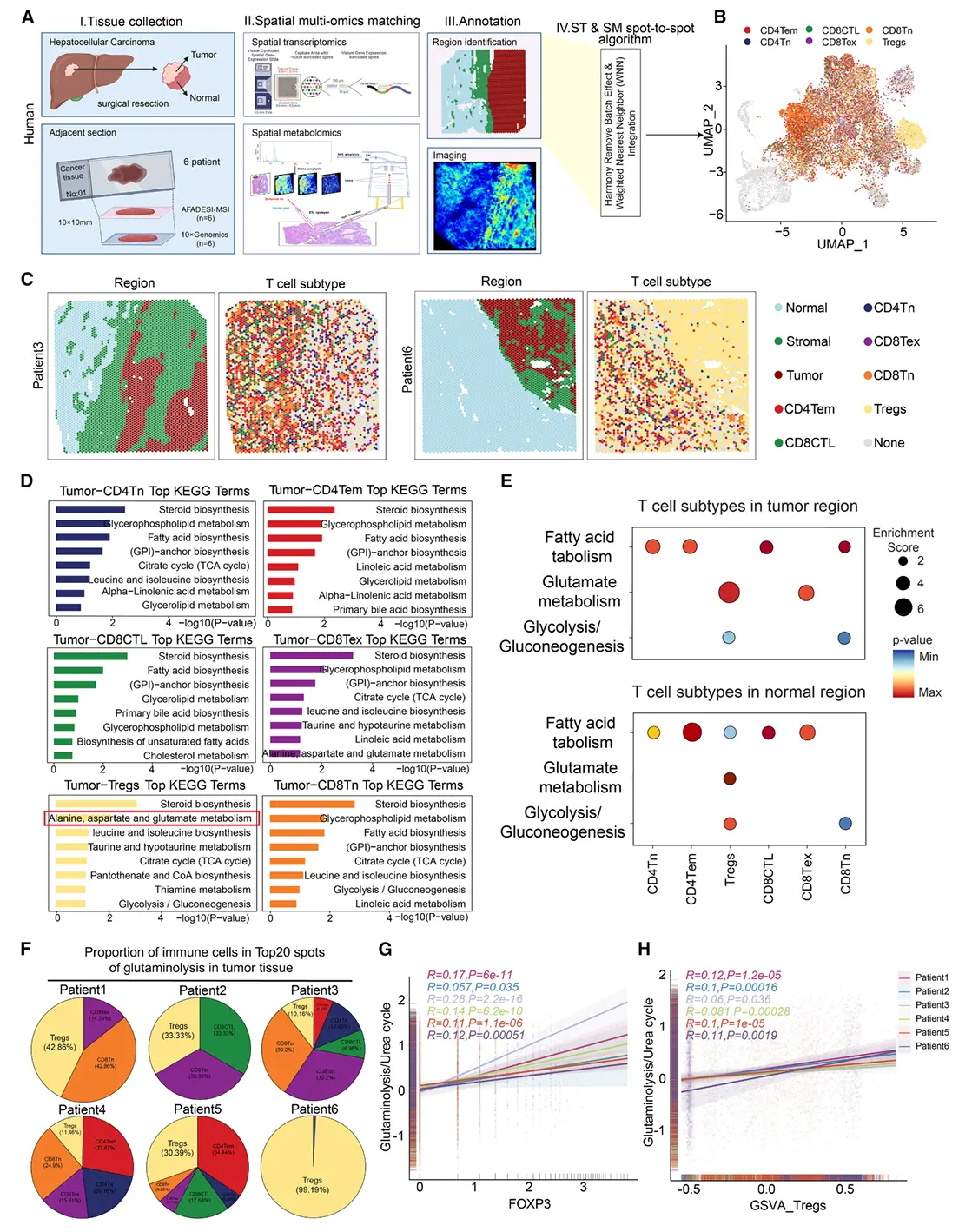
结果1、高谷氨酰胺分解与低尿素循环活性是调节性T细胞富集肿瘤亚区的关键代谢特征
为实现空间转录组与空间代谢组数据整合,将空间转录组的条形码空间信息与空间代谢组的像素点信息转换为统一空间标识符,并通过累加每个转录组点位对应像素点的离子强度建立关联,最终获得与空间转录组对齐的重整合空间代谢组数据。
在谷氨酰胺分解活跃的亚区内存在明显的Tregs富集。
谷氨酰胺分解(而非糖酵解)是肿瘤细胞的主要能量来源,且与免疫逃逸机制密切相关。
仅当同时具备"高谷氨酰胺分解-低尿素循环活性"的代谢特征时(而非任一通路单独作用),才与FOXP3⁺ Tregs的丰度及功能呈强相关性。
氨是谷氨酰胺分解的主要副产物,主要通过尿素循环代谢。因此,尿素循环活性降低会导致氨积累,而活性增强则促进解毒。我们由此推测氨可能促进Tregs积累。
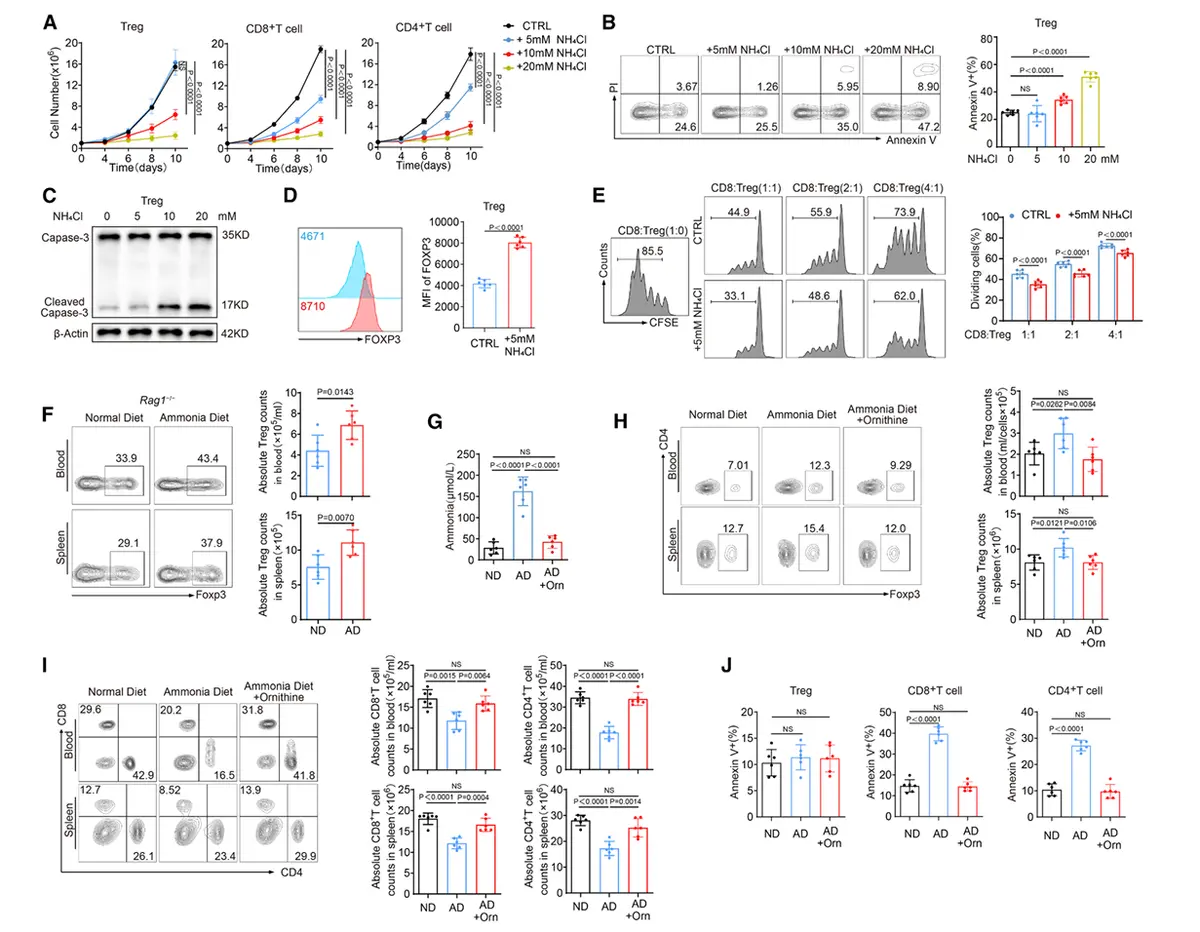
结果2、调节性T细胞抵抗氨诱导的凋亡而CD8⁺与CD4⁺ T细胞不具备此能力
体外分离健康人外周血单个核细胞来源的调节性T细胞、CD8⁺ T细胞与CD4⁺ T细胞,并暴露于不同浓度氯化铵处理环境。模拟瘤内水平的氨浓度(5 mM)未影响调节性T细胞数量,但显著抑制CD8⁺与CD4⁺ T细胞增殖。更高氨浓度(10 mM与20 mM)则抑制所有T细胞亚群,且对CD8⁺及CD4⁺ T细胞的抑制作用更为显著。
在低浓度与高浓度氨环境下,调节性T细胞死亡率均低于CD8⁺与CD4⁺ T细胞。低氨条件下,调节性T细胞能维持稳定的胞质pH值、较低的胞内氨水平,并保持溶酶体与线粒体结构完整;而高氨暴露则会破坏这种抵抗能力。流式细胞术进一步显示,低氨水平下仅调节性T细胞能抵抗凋亡,CD8⁺与CD4⁺ T细胞则不能。
氨能选择性诱导CD4⁺与CD8⁺ T细胞凋亡,同时保护调节性T细胞并上调其PD-1与CTLA-4表达。
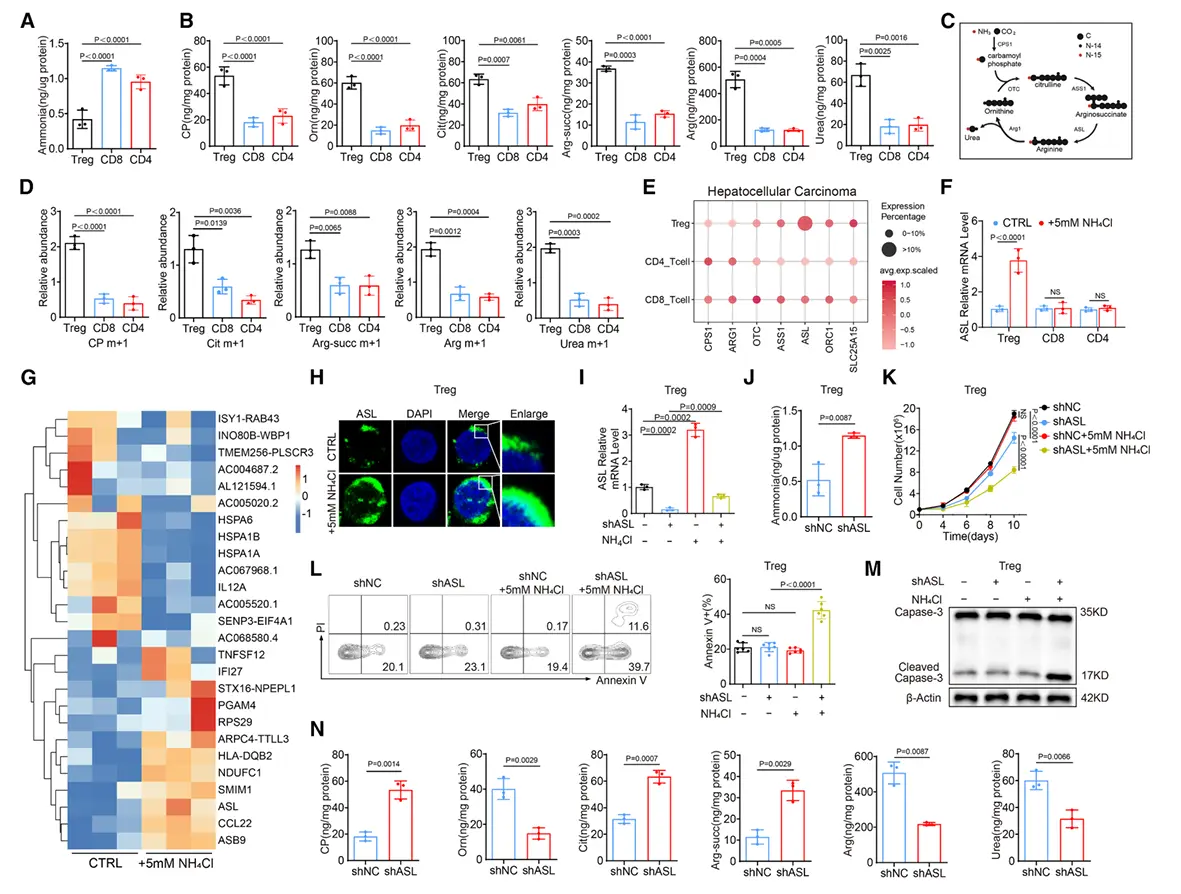
结果3、氨暴露通过上调精氨琥珀酸裂解酶表达激活调节性T细胞尿素循环
在富氨环境中调节性T细胞展现出更强的尿素循环活性。
对已发表的人肝细胞癌单细胞RNA测序数据进行分析,发现肿瘤浸润调节性T细胞特异性高表达尿素循环相关酶,尤其是精氨琥珀酸裂解酶水平显著高于其他T细胞;相比之下,外周血中各T细胞亚群的ASL表达量相近。这提示氨暴露可能上调调节性T细胞中ASL表达。
ASL缺失使调节性T细胞对氨诱导凋亡敏感,同时减缓肿瘤进展。这种调节性T细胞介导的免疫抑制减弱可能改善治疗效果。更重要的是,调节性T细胞中ASL缺陷可增强抗PD-1疗法的响应。
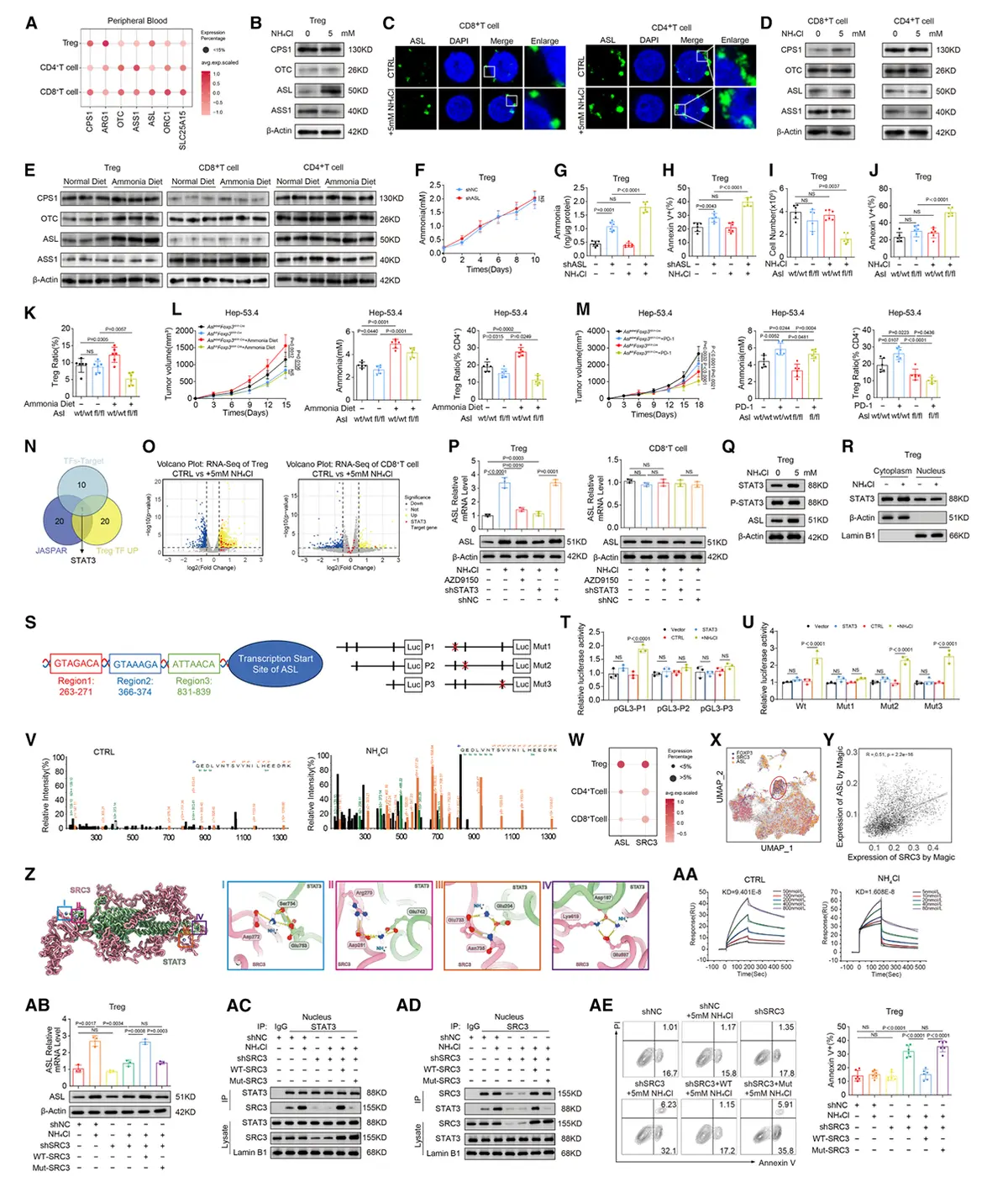
氨通过SRC3介导的STAT3活化增强调节性T细胞中ASL转录
氨处理能强烈诱导对照调节性T细胞的ASL转录,增强核内STAT3-SRC3复合物形成,并减少细胞凋亡;这些效应在SRC3敲低细胞中消失,且仅能被野生型SRC3恢复。综上,氨通过促进SRC3-STAT3相互作用驱动ASL转录,而SRC3在调节性T细胞中的选择性富集(文献报道其对于调节性T细胞功能具有重要促进作用),构成了该细胞在肿瘤微环境中感知与适应氨应激的特有能力基础。
结果4、氨通过FOXP3驱动的精胺合酶表达促进调节性T细胞氧化磷酸化
在富氨环境中,调节性T细胞展现出增强的功能活性。
电镜观察进一步表明,氯化铵处理的调节性T细胞能维持线粒体结构完整性,并伴有ATP产量增加与膜电位升高。此外,在不同pH条件下无论是否添加氨,调节性T细胞的氧化磷酸化水平均保持稳定。还证实氨能在转录、翻译及翻译后水平上调FOXP3表达,同时增强调节性T细胞抑制分子表达并稳定其功能,这些效应均可被氧化磷酸化抑制剂部分阻断。
基于JASPAR与PROMO数据库分析,调节性T细胞特异性转录因子FOXP3被鉴定为SMS的潜在调控因子。对SMS启动子分析发现五个高可信度的FOXP3结合区域,定点缺失与突变实验进一步证实区域2和区域5对FOXP3介导的SMS上调具有关键作用。染色质免疫共沉淀与电泳迁移率变动实验验证了FOXP3与SMS启动子的直接结合。这些结果确立了FOXP3作为转录激活因子驱动SMS表达,从而增强调节性T细胞特异性的氨诱导氧化磷酸化过程。
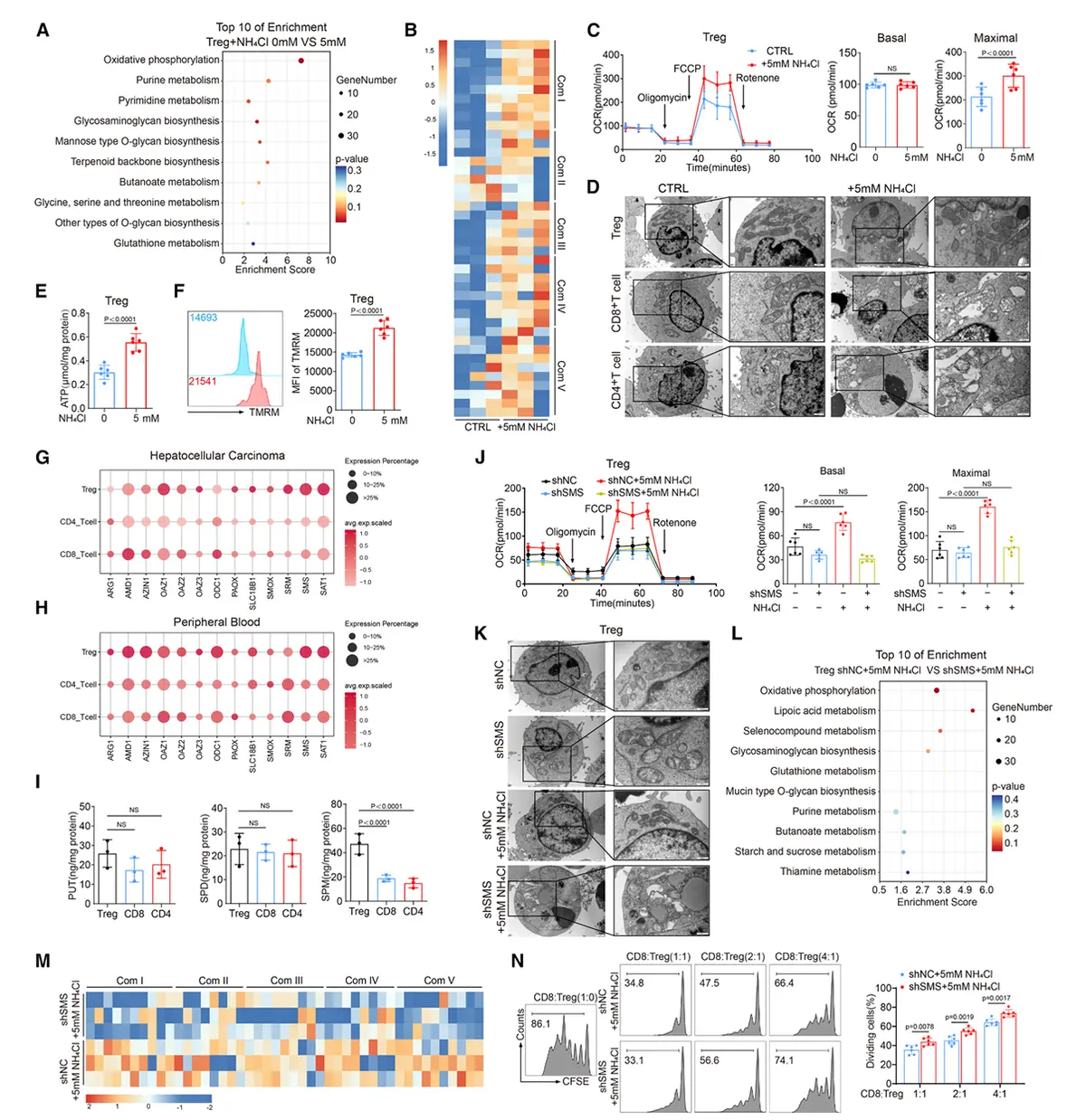
结果5、精胺通过PPARγ增强调节性T细胞氧化磷酸化与免疫抑制能力
精胺是氨诱导Tregs代谢增强的关键代谢物
精胺合酶(SMS)催化亚精胺转化为精胺,该过程是氨促进Tregs氧化磷酸化(OXPHOS)所必需的。
外源性精胺无法直接进入细胞,但通过补充其前体亚精胺(可在细胞内转化为精胺)可显著增强Tregs的OXPHOS能力。
抑制精胺降解(如过表达限速酶SAT1)会降低细胞内精胺水平并削弱OXPHOS,进一步证实精胺是驱动氨依赖性代谢增强的核心代谢物。
精胺通过直接靶向PPARγ发挥作用
基于精胺衍生探针(SPM-DA)的化学生物学策略,结合蛋白质组学与转录因子分析,鉴定出PPARγ是精胺在Tregs中的直接作用靶点。
精胺与PPARγ结合具有高度特异性,且呈剂量依赖性。
PPARγ的缺失或敲低可完全阻断氨(或精胺)诱导的OXPHOS增强、线粒体功能改善及免疫抑制功能提升。
PPARγ是氨-精胺轴调控Tregs功能的核心枢纽
氨处理不改变PPARγ表达水平,但通过精胺介导的PPARγ活化驱动下游代谢与功能重编程。
在动物模型中,Tregs特异性敲除PPARγ可逆转氨对肿瘤进展的促进作用,并抑制Tregs在肿瘤微环境中的积累。
这表明PPARγ是连接氨代谢产物精胺与Tregs功能增强的关键信号节点。
机制总结:肿瘤微环境中累积的氨 → 通过FOXP3上调SMS表达 → 促进亚精胺向精胺转化 → 精胺直接结合并激活PPARγ → 增强线粒体OXPHOS、维持代谢适应性 → 最终强化Tregs的免疫抑制功能与肿瘤内生存优势。
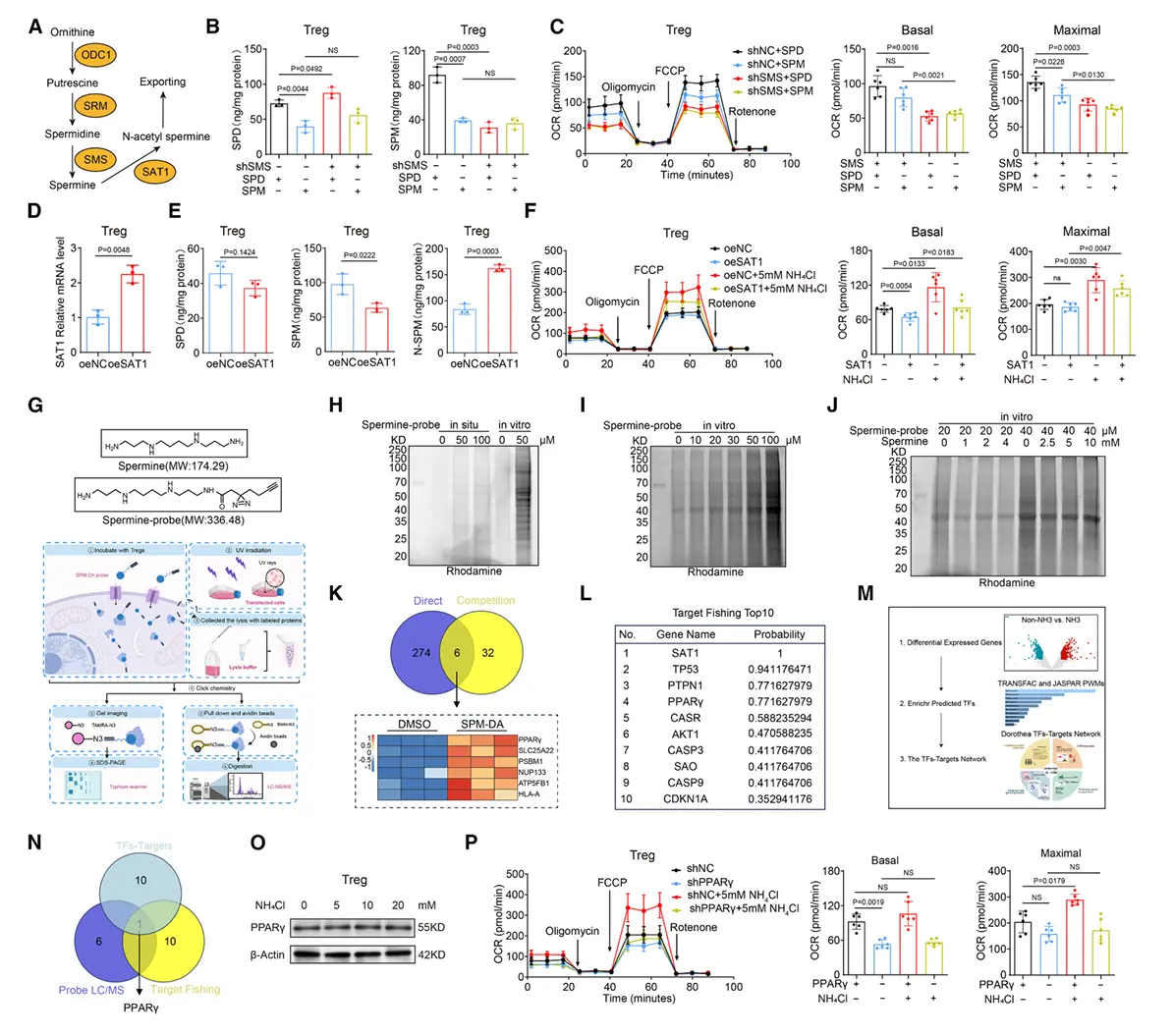
结果6、精胺直接结合PPARγ调控线粒体复合物组装与Tregs氧化磷酸化
证实精胺与PPARγ直接结合
结合亲和力验证
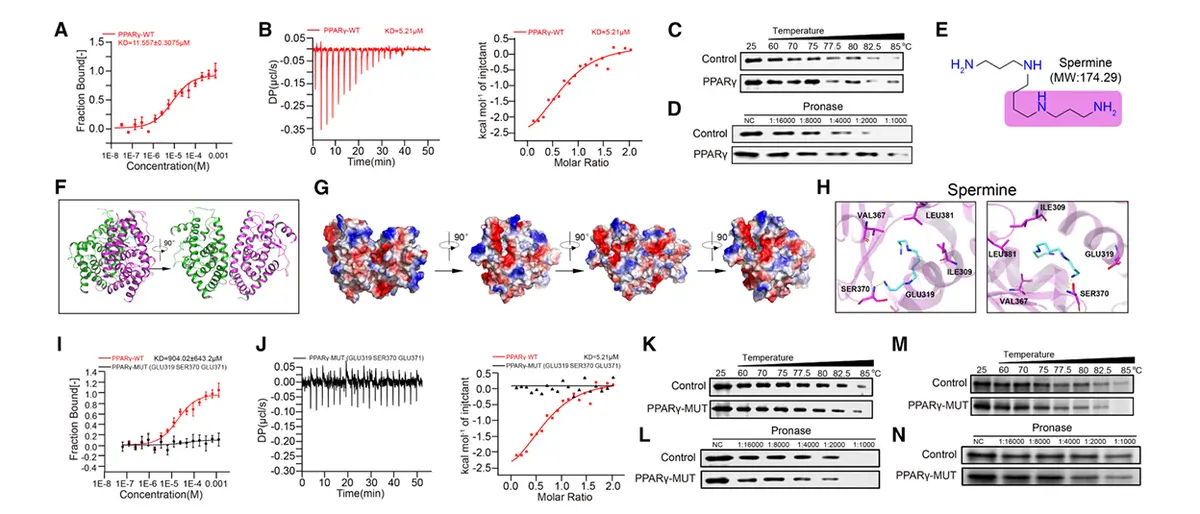
微量热泳动实验与等温滴定量热法测定结合常数分别为 KD = 11.557 ± 3.075 μM 与 5.21 μM,证实两者存在特异性相互作用。
结合稳定性验证
细胞热迁移实验与药物亲和反应靶点稳定性实验显示,精胺能增强PPARγ的热稳定性并保护其免受蛋白酶降解。
结构互作机制解析
分子对接与晶体结构分析表明,精胺通过静电作用与氢键与PPARγ的 Glu319、Ser370、Glu371 残基结合。
将上述关键残基突变为丙氨酸(PPARγ-MUT)可完全消除精胺的结合能力。
精胺-PPARγ互作的功能学意义
突变体功能丧失
在 PparγMut Foxp3YFP-Cre 小鼠中,氨处理无法增强Tregs的免疫抑制功能与氧化磷酸化水平,线粒体形态、ATP产量及膜电位均未见改善。
调控线粒体复合物表达
精胺通过PPARγ转录活性调控多个线粒体呼吸链复合物亚基的表达,包括:
- 复合物I:NDUFA1、NDUFC1
- 复合物II:SDHAF1、SDHC
- 复合物III:UQCRQ、UQCRH
- 复合物IV:COX6C、COX5B
- 复合物V:ATP1B1、ATP1B3
双荧光素酶报告基因与染色质免疫共沉淀实验证实,精胺-PPARγ结合可直接促进NDUFC1等靶基因的转录。
ASL与PPARγ通路的贡献比较
ASL缺失(影响氨解毒)比 PPARγ突变(影响精胺信号)对肿瘤生长的抑制效果更显著,表现为更明显的肿瘤体积减小、瘤内氨水平下降及Tregs积累减少。
机制在不同癌种中的普适性
在肺腺癌、结肠腺癌及胆管癌的公共单细胞数据集中,高谷氨酰胺酶肿瘤内的Tregs均呈现:
更高的免疫抑制评分
ASL与PPARγ活化增强
氧化磷酸化通路富集
这表明"氨解毒→精胺积累→PPARγ激活→OXPHOS增强"是Tregs在多种肿瘤微环境中适应代谢压力的保守机制。
机制整合:肿瘤氨积累 → Tregs通过ASL介导的尿素循环解毒 → 生成精胺 → 直接结合并稳定PPARγ → 激活线粒体呼吸链基因转录 → 促进线粒体复合物组装与OXPHOS → 增强Tregs存活与免疫抑制功能。
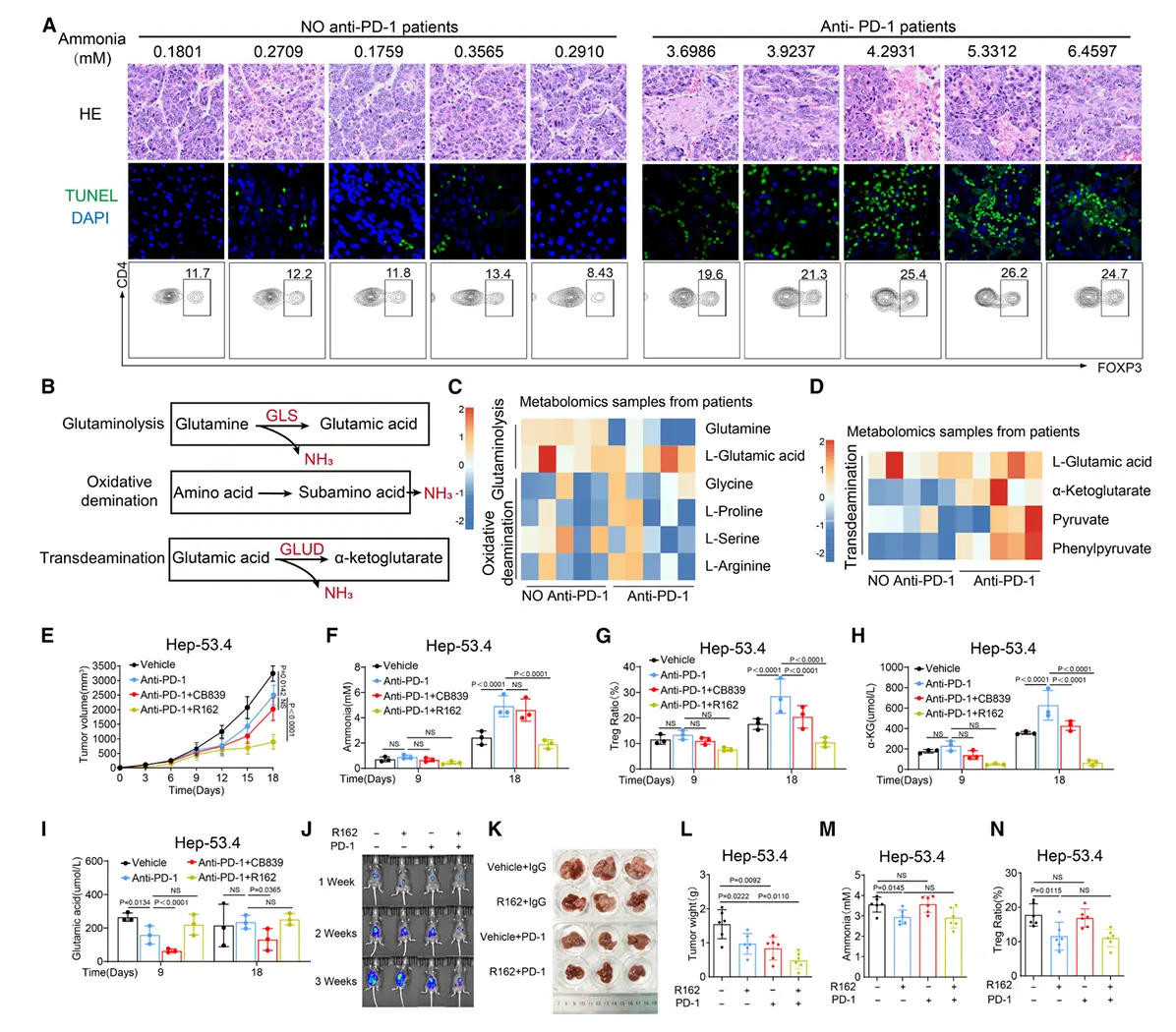
结果7、抗PD-1疗法诱导的细胞死亡产氨增强Tregs并导致免疫治疗耐药
临床与实验证据:抗PD-1治疗伴随氨积累与Tregs增加
接受抗PD-1治疗后进行转化手术的肝细胞癌患者样本中,肿瘤组织氨水平显著升高,同时肿瘤浸润Tregs比例增加。
多种肿瘤细胞系(包括Hep-53.4)在诱导凋亡后,培养上清中氨浓度明显上升。
机制解析:细胞死亡相关氨的来源是转脱氨作用
代谢组学分析
肿瘤细胞凋亡时,转脱氨作用(transdeamination)被显著激活,而非谷氨酰胺分解或氧化脱氨。
临床抗PD-1治疗标本中也检测到转脱氨通路活性增强。
关键酶验证
使用谷氨酰胺缺失培养基或GLS1抑制剂不影响凋亡相关的氨生成,排除谷氨酰胺分解为主要来源。
GLUD(谷氨酸脱氢酶)抑制剂可显著减少氨产生,证实谷氨酸经转脱氨生成α-酮戊二酸是凋亡诱导氨的主要来源。
动物模型验证:靶向GLUD可逆转PD-1耐药
耐药模型中的代谢变化
皮下Hep-53.4肿瘤模型中,早期抗PD-1治疗可延缓肿瘤生长,但随后出现耐药且伴随:
- 瘤内氨水平升高
- Tregs比例增加
- α-酮戊二酸积累(反映转脱氨激活)
联合治疗的效果
GLS1抑制剂无法逆转PD-1耐药或降低氨水平。
GLUD抑制剂(R162) 可:
- 有效控制肿瘤再生
- 降低瘤内氨浓度
- 减少Tregs浸润
- 提高CD8⁺ T细胞比例与功能,减少其凋亡
在原位肝癌、HTVI-HCC及MASH-HCC模型中均观察到类似疗效。
治疗潜力:GLUD1抑制增强免疫疗效
单用GLUD1抑制剂已在多种肿瘤模型中显示抗肿瘤活性(抑制Tregs积累)。
GLUD1抑制剂联合抗PD-1治疗可提升抗肿瘤效果(增强幅度达33.1%--63.2%),尤其在免疫"热"肿瘤中效果显著。
初步数据支持靶向GLUD1具有临床转化潜力。
机制总结
抗PD-1治疗 → 肿瘤细胞凋亡 → 激活转脱氨作用(GLUD介导)→ 氨生成增加 → 促进Tregs扩增/功能增强 → 抑制CD8⁺ T细胞 → 免疫治疗耐药
↓
联合GLUD抑制剂 → 阻断氨生成 → 减少Tregs → 恢复CD8⁺ T细胞功能 → 逆转耐药
最后,来看看方法
