肿瘤如何通过密集的血管网获取养分并完成逃逸,其血管内部的每个细胞究竟扮演什么角色?
2024年7月10日,《Nature》在线发表了由重庆大学三峡医院尹明珠团队领衔的研究,该研究构建了一个包含31种癌症类型、约20万个细胞的肿瘤血管单细胞图谱,揭示了肿瘤血管生成起源于静脉内皮细胞的动态过程。今天我们就来拆解一下这篇生信文章:Tumour vasculature at single-cell resolution。

研究概述
肿瘤进展和转移高度依赖血管提供的氧气和营养物质。研究团队生成了涵盖372名捐赠者的全面肿瘤血管系统单细胞图谱,包含内皮细胞和壁细胞共计183,977个。研究发现,肿瘤血管生成是由静脉内皮细胞启动并向动脉内皮细胞方向延伸的。研究鉴定了与预后不良及抗VEGF疗法响应相关的
TipSI细胞,并发现了淋巴内皮细胞的两条分化路径以及由内质网应激驱动的促血管生成周细胞亚群。该研究为理解肿瘤血管复杂性及优化抗血管治疗提供了系统证据。
实验设计
研究整合了本中心及58个公开数据集的437个样本,涵盖31种癌症类型。研究通过in silico分选提取血管内皮细胞(ECs)和壁细胞(MCs)。为了验证转录组分析结果,研究团队构建了肿瘤细胞异种移植斑马鱼模型观察血管出芽,并结合多色免疫组化(mIHC)以及空间转录组测序技术,在人类肿瘤组织样本中验证关键细胞亚群的分布与相互作用。
研究结果
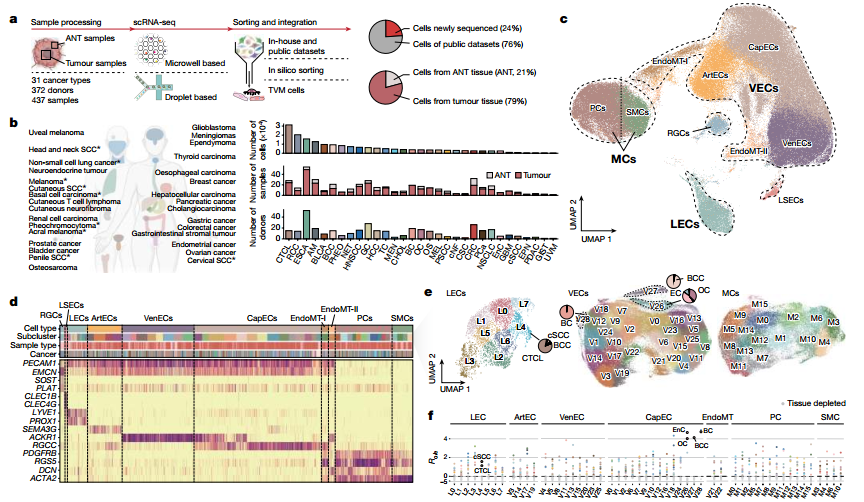 图1 :研究整合生成了跨31种癌症类型的肿瘤血管单细胞图谱,并将其划分为内皮细胞、壁细胞等29个血管内皮、8个淋巴内皮及16个壁细胞亚群。
图1 :研究整合生成了跨31种癌症类型的肿瘤血管单细胞图谱,并将其划分为内皮细胞、壁细胞等29个血管内皮、8个淋巴内皮及16个壁细胞亚群。
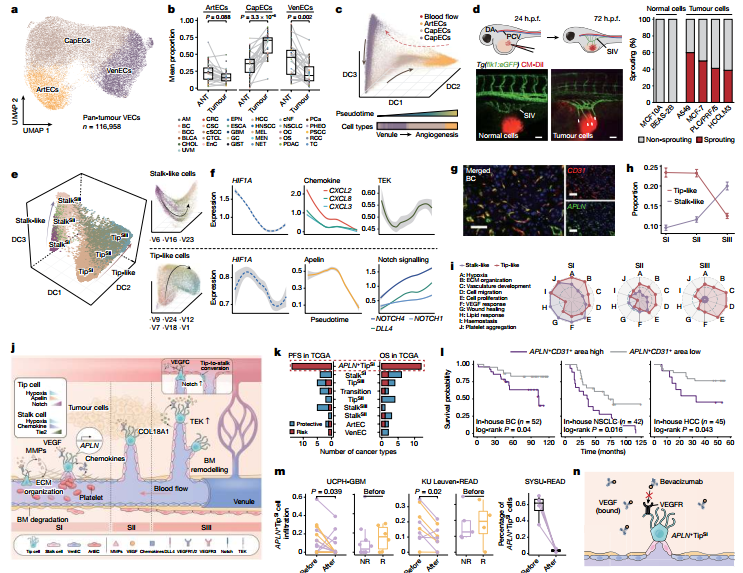 图2 :轨迹推断与斑马鱼实验共同证明肿瘤血管生成起源于静脉内皮细胞,并识别出具有预后预测价值的 TipSI细胞。
图2 :轨迹推断与斑马鱼实验共同证明肿瘤血管生成起源于静脉内皮细胞,并识别出具有预后预测价值的 TipSI细胞。
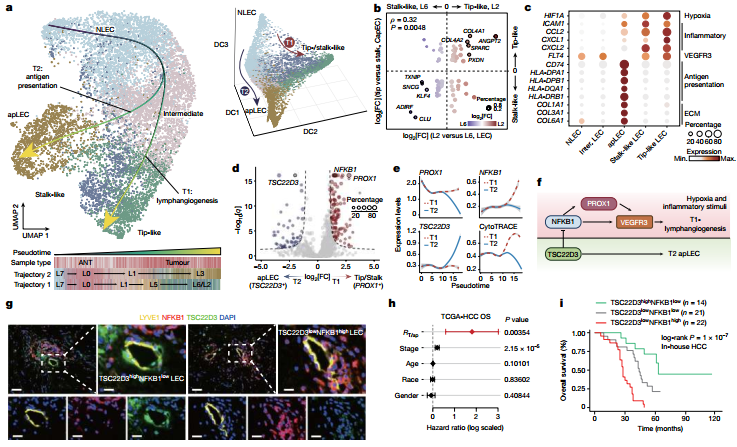 图3 :淋巴内皮细胞存在两条分化谱系,分别负责肿瘤淋巴管生成和抗原提呈,且具有不同的临床预后意义。
图3 :淋巴内皮细胞存在两条分化谱系,分别负责肿瘤淋巴管生成和抗原提呈,且具有不同的临床预后意义。
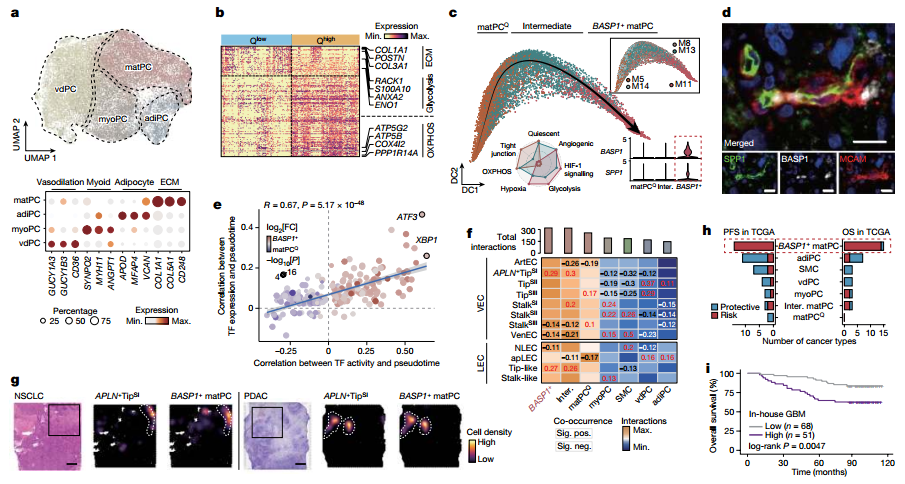 图4 :周细胞中的内质网应激促进了 基质产生型周细胞的分化,该亚群与新生血管空间相邻并促进血管生成。
图4 :周细胞中的内质网应激促进了 基质产生型周细胞的分化,该亚群与新生血管空间相邻并促进血管生成。
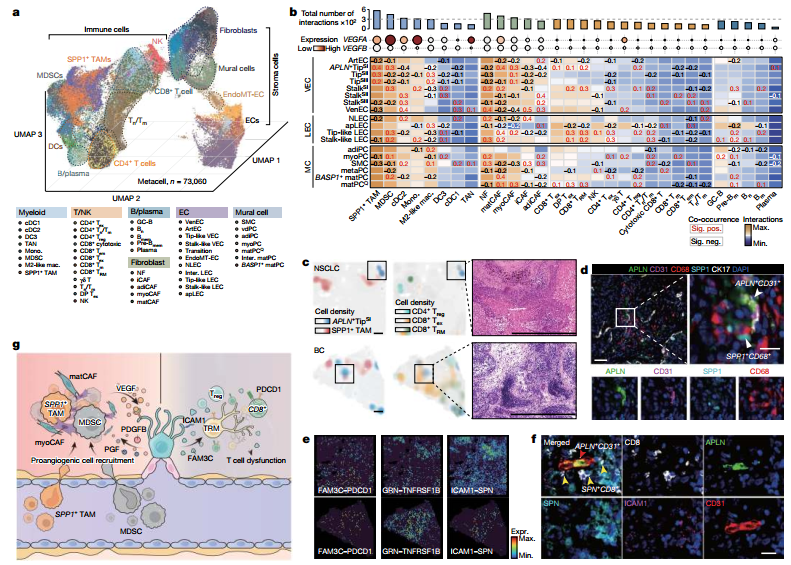 图5 :血管细胞与肿瘤微环境细胞通过复杂的配受体相互作用,共同构建了一个支持血管生成且具有免疫抑制特性的微环境。
图5 :血管细胞与肿瘤微环境细胞通过复杂的配受体相互作用,共同构建了一个支持血管生成且具有免疫抑制特性的微环境。
数据分析
生信分析
涉及技术
该研究涉及单细胞转录组测序(scRNA-seq)、空间转录组测序、多色免疫组化(mIHC)和染色质免疫共沉淀测序(ChIP-seq)等组学技术。
单细胞RNA测序分析
-
- 数据预处理:使用CellRanger、BD Rhapsody Sequence Analysis Pipeline等工具处理不同平台测序数据,比对至GRCh38基因组。
-
- 质量控制:过滤线粒体基因占比>30%的细胞,去除双细胞,校正细胞周期、供者等混杂因素。
-
- 数据整合与批次校正:通过Harmony、CSS方法整合多平台数据,降低批次效应。
-
- 细胞聚类与注释:基于典型标志物基因(如PECAM1、RGS5)注释细胞类型及亚型,进行无监督聚类得到亚群。
-
- 功能分析:通过AUCell计算基因集活性,SOM分析鉴定肿瘤血管生成相关功能特征,NicheNet、CellPhoneDB分析细胞间通讯。
-
- 轨迹推断:使用Diffusion Map、Slingshot等多种算法构建细胞分化轨迹,CytoTRACE评估细胞分化状态。
空间转录组分析流程
-
- 数据处理:通过Space Ranger pipeline处理原始数据,获取空间基因表达矩阵。
-
- 细胞类型反卷积:应用CARD方法结合单细胞参考数据,推断空间位置的细胞类型组成。
-
- 共定位分析:计算配体-受体对的表达强度,分析功能相关细胞的空间分布关系。
其他组学分析
-
- ChIP-seq:获取XBP1、ATF3等转录因子的结合区域数据,分析其对下游VEGF基因的调控作用。
-
- mIHC:通过PhenoImager HT获取图像数据,ImageJ定量分析标志物表达,病理学家验证结果。
组学联合分析
整合scRNA-seq的细胞亚群信息与空间转录组的位置信息,明确功能细胞亚群的空间分布;结合ChIP-seq的转录因子结合数据,揭示内质网应激调控促血管生成细胞表型的分子机制。
统计分析
-
- 组间比较:采用Mann-Whitney U检验比较两组数据差异,Kruskal-Wallis检验分析多组数据差异。
-
- 生存分析:通过Cox比例风险模型评估细胞亚群浸润水平与预后的关联,Log-rank检验验证生存曲线差异。
-
- 相关性分析:使用Spearman相关系数分析细胞亚群比例及基因表达的相关性。
-
- 富集分析:基于累积超几何分布计算功能富集的P值,校正多重检验误差。
统计分析
研究采用双侧Mann-Whitney U检验进行两组间比较,使用Kruskal-Wallis检验处理多组间差异。生存分析采用log-rank检验和Cox比例风险模型,以评估细胞浸润水平与整体生存期(OS)或无进展生存期(PFS)的关联,并校正年龄、性别、种族和临床阶段等混杂因素。相关性分析则使用Spearman相关系数分析细胞亚群比例及基因表达。富集分析则基于累积超几何分布计算功能富集的P值,校正多重检验误差。
总结
研究意义
本研究构建了全面的肿瘤血管单细胞图谱,阐明了肿瘤血管生成的细胞起源和分化规律,鉴定出APLN+TipSI细胞、BASP1+周细胞等具有临床价值的靶点,为抗血管生成治疗的患者筛选、疗效预测提供了新依据,同时为肿瘤血管生物学的深入研究提供了数据资源和分析框架。
文章复现
这篇文章的原始数据和生信分析代码都公开了,非常全面。
原始数据及代码仓库地址如下:
-
• 原始数据: Genome Sequence Archive(国家基因组科学数据中心),BioProject ID:PRJCA018695
-
• 生信分析代码:GitHub仓库:https://github.com/bio-Pixel/panVC
-
• 生信分析代码:Zenodo仓库:https://zenodo.org/records/11188740
推荐阅读
中国银河生信云平台(UseGalaxy.cn)致力于零代码生信分析。平台拥有海量计算资源、3000 多个生信工具和数十条生信流程,并且为用户提供 200G 免费存储空间。进群交流请先加 usegalaxy 为好友。
一文学会从测序数据到构建系统发育树:超全面的详细步骤与软件指南



推荐课程
我们还为进阶用户提供高质量培训课程,欢迎参加: