引言
在基因组学研究中,一个基本问题是:基因组大小与编码基因(CDS)数量之间存在怎样的关系? 不同来源的基因组------如单细胞基因组(SAG)、宏基因组组装基因组(MAG)和完整测序基因组(WGS)------可能表现出不同的标度律。为了直观地探索这种关系,我们常常需要将数据可视化,并辅以统计检验。
本文分享一段完整的Python代码,它能够:
- 读取或模拟基因组大小与CDS数量的数据;
- 在对数空间进行线性拟合,计算各组样本的残差;
- 绘制联合分布图(散点图 + 边缘密度),并叠加回归线;
- 在主图中嵌入一个残差箱线图,比较不同基因组类型偏离总体趋势的程度;
- 自动添加统计显著性标注(Mann‑Whitney U检验)。
- 最终输出一张信息丰富、可直接用于出版的矢量图(PDF)。所有参数集中配置,方便修改和复用。
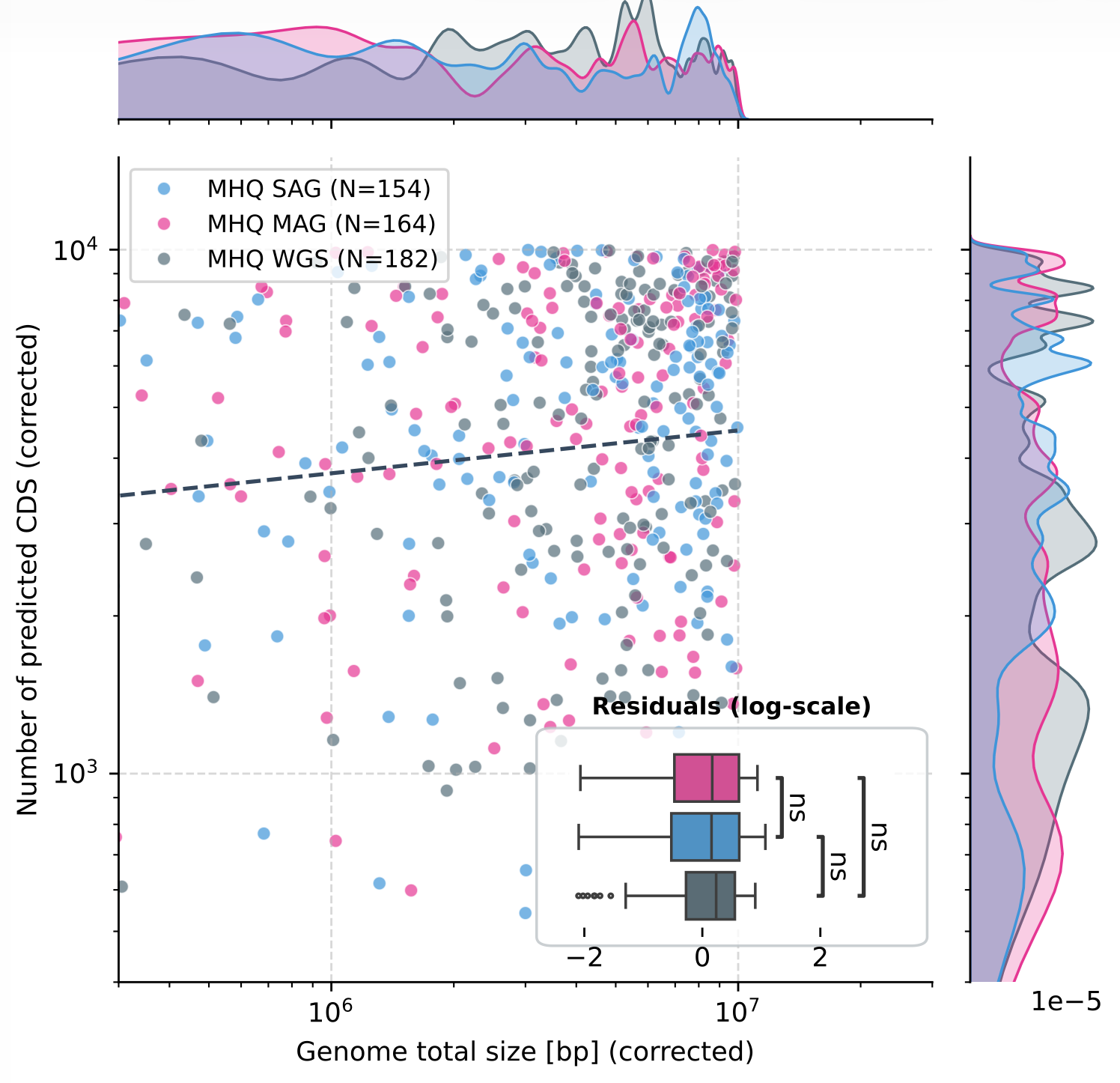
1. 数据准备与预处理
1.1 数据来源
代码支持两种数据来源:
- 真实数据:从 data.csv 读取,要求包含 length(基因组长度)、number_of_cds(CDS数量)和 type(SAG/MAG/WGS)三列。
- 模拟数据:若文件不存在,自动生成500个样本,用于测试。
1.2 对数变换与线性拟合
生物学数据常遵循幂律分布y=a⋅xby= a \cdot x^by=a⋅xb,取对数后变为线性关系:
log(CDS)=b⋅log(length)+log(a)log(CDS)=b⋅log(length)+log(a)log(CDS)=b⋅log(length)+log(a)
我们使用 scipy.optimize.curve_fit 对全体数据(不分类型)进行最小二乘拟合,得到斜率bbb和截距log(a)log(a)log(a)。这一趋势线代表了基因组大小与CDS数量的总体关系。
1.3 残差计算
残差定义为实际值的对数与拟合值的对数之差:
residual=log(CDSobs)−log(CDSfit)residual=log(CDS_{obs} )−log(CDS_{fit})residual=log(CDSobs)−log(CDSfit)
正值表示该基因组比总体趋势预测的CDS更多,负值则更少。后续通过比较不同组的残差,可以判断哪类基因组系统性偏离总体趋势。
python
# 核心拟合代码
def linear_func(x, a, b):
return a * x + b
log_len = np.log(df["length"])
log_cds = np.log(df["number_of_cds"])
popt, _ = sciopt.curve_fit(linear_func, log_len, log_cds)
df["residuals_log"] = log_cds - linear_func(log_len, *popt)2. 主图:联合分布散点图 + 边缘密度
我们使用 seaborn.jointplot 绘制主图,它能同时展示散点分布和两个变量的边缘密度分布。
2.1 散点图设置
- 横轴为基因组大小(length),纵轴为CDS数量(number_of_cds),双对数坐标。
- 颜色按 type 区分,使用自定义调色板(蓝-粉-灰)。
- 散点添加白色边缘,增强可读性。
2.2 回归线
将拟合得到的幂律曲线绘制在原始坐标空间:
CDSfit=eb⋅log(length)+log(a)CDS_{fit} =e^{b⋅log(length)+log(a)}CDSfit=eb⋅log(length)+log(a)
2.3 边缘密度
通过 marginal_kws 控制核密度估计的带宽和填充,让分布形态一目了然。
2.4 图例增强
自动计算每类样本的数量,在图例中显示为 MHQ SAG (N=123) 形式,便于读者了解样本量。
python
g = sns.jointplot(data=df, x="length", y="number_of_cds", hue="type",
kind="scatter", alpha=0.7, height=6,
palette=CONFIG["colors"],
joint_kws={"s": 25, "edgecolor": "w", ...},
marginal_kws={"bw_adjust": 0.2, "fill": True})3. 内嵌箱线图:残差分布与显著性检验
为了在不占用额外空间的情况下展示组间差异,我们在主图右下角嵌入一个水平箱线图,用于比较三类样本的对数残差分布。
3.1 创建内嵌坐标轴
使用 mpl_toolkits.axes_grid1.inset_locator.inset_axes 在主图坐标轴内部开辟一个小区域,通过 bbox_to_anchor 精确定位。
3.2 绘制箱线图
- 横轴为残差值,纵轴为类型(WGS、MAG、SAG),水平方向绘制。
- 颜色顺序与主图一致:灰(WGS)、粉(MAG)、蓝(SAG)。
- 隐藏默认的刻度线和边框,使内嵌图更干净。
3.3 统计标注
使用 statannotations.Annotator 自动进行两两 Mann‑Whitney U 检验,并在图上添加星号表示显著性(* p<0.05, ** p<0.01, *** p<0.001)。
3.4 添加背景框
通过 FancyBboxPatch 绘制一个带圆角的半透明白色背景,让内嵌图从主图中突出。
python
ax_ins = inset_axes(g.ax_joint, width="40%", height="22%", loc="lower right", ...)
sns.boxplot(data=df, x="residuals_log", y="type", hue="type",
order=["WGS", "MAG", "SAG"], palette=box_colors, ax=ax_ins)
annotator = Annotator(ax_ins, [("WGS","MAG"), ("WGS","SAG"), ("MAG","SAG")], ...)
annotator.configure(test="Mann-Whitney", text_format="star")
annotator.apply_and_annotate()4. 完整代码
python
# ==========================================
# 全基因组规模 vs CDS 数量分析(完整脚本)
# ==========================================
import random
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib as mpl
import scipy.optimize as sciopt
import seaborn as sns
from mpl_toolkits.axes_grid1.inset_locator import inset_axes
from statannotations.Annotator import Annotator
from matplotlib.patches import FancyBboxPatch
# ----------------------------------------------------------
# 1. 全局配置:在此修改参数,全局生效
# ----------------------------------------------------------
random.seed(10)
np.random.seed(10)
CONFIG = {
"seed": 10,
"input_file": "data.csv", # 真实数据路径,不存在时自动生成模拟数据
"colors": ["#3498db", "#e84393", "#546e7a"], # 蓝, 粉, 灰
"hue_order": ["SAG", "MAG", "WGS"],
"plot_limits": {
"xlim": (3e5, 3e7), # 3e5 ~ 3e7 bp
"ylim": (400, 15000), # CDS 数量范围
"xlabel": "Genome total size [bp] (corrected)",
"ylabel": "Number of predicted CDS (corrected)"
}
}
# ----------------------------------------------------------
# 2. 数据处理:读取 + 对数空间线性拟合 + 残差计算
# ----------------------------------------------------------
def prepare_data(file_path):
"""
读取原始数据,进行对数空间的线性拟合,并计算残差。
若文件不存在,则生成模拟数据用于测试。
Parameters
----------
file_path : str
CSV 文件路径。
Returns
-------
df : pandas.DataFrame
包含原始数据、拟合值及对数残差的数据框。
popt : numpy.ndarray
拟合参数 [斜率, 截距]。
"""
try:
df = pd.read_csv(file_path, index_col=0)
print(f"成功加载数据:{file_path}")
except FileNotFoundError:
print(f"文件 {file_path} 不存在,正在生成模拟数据(500个样本)...")
data = {
"length": np.random.uniform(1e5, 1e7, 500),
"number_of_cds": np.random.uniform(500, 10000, 500),
"type": np.random.choice(CONFIG["hue_order"], 500)
}
df = pd.DataFrame(data)
# 对数转换------生物数据通常服从幂律分布
log_length = np.log(df["length"])
log_cds = np.log(df["number_of_cds"])
# 线性模型:log(cds) = a * log(length) + b
def linear_func(x, a, b):
return a * x + b
# 最小二乘拟合
popt, _ = sciopt.curve_fit(linear_func, log_length, log_cds)
# 计算拟合值及对数残差
df["log_length"] = log_length
df["log_cds"] = log_cds
df["cds_fit"] = np.exp(linear_func(log_length, *popt))
df["residuals_log"] = log_cds - np.log(df["cds_fit"])
return df, popt
# ----------------------------------------------------------
# 3. 主绘图:联合分布散点图 + 边缘密度 + 回归线
# ----------------------------------------------------------
def plot_main_joint(df, popt):
"""
绘制主图:联合分布散点图(对数坐标)+ 边缘密度 + 对数空间回归线。
Parameters
----------
df : pandas.DataFrame
包含 'length', 'number_of_cds', 'type' 及对数残差的数据框。
popt : numpy.ndarray
拟合参数 [斜率, 截距],用于绘制回归线。
Returns
-------
g : seaborn.JointGrid
JointGrid 对象,可继续添加子图或修改样式。
"""
g = sns.jointplot(
data=df,
x="length", y="number_of_cds", hue="type",
kind="scatter", alpha=0.7, height=6,
palette=CONFIG["colors"],
hue_order=CONFIG["hue_order"],
joint_kws={"s": 25, "edgecolor": "w", "linewidth": 0.5},
marginal_kws={"bw_adjust": 0.2, "fill": True}
)
# 消除顶部边缘图 Y 轴的科学计数法
g.ax_marg_x.ticklabel_format(axis="y", style="plain")
# 设置坐标轴为对数刻度,并应用全局绘图范围
g.ax_joint.set(xscale="log", yscale="log", **CONFIG["plot_limits"])
g.ax_joint.grid(True, which="major", linestyle="--", alpha=0.5, zorder=0)
# 在原始坐标空间绘制回归线
x_log_range = np.linspace(df["log_length"].min(), df["log_length"].max(), 100)
x_range = np.exp(x_log_range)
y_fit = np.exp(popt[0] * np.log(x_range) + popt[1])
g.ax_joint.plot(x_range, y_fit, color="#34495e", linestyle="--", lw=1.5, zorder=5)
# 图例:添加样本量 (N=xxx)
type_counts = df["type"].value_counts()
handles, labels = g.ax_joint.get_legend_handles_labels()
new_labels = [
f"MHQ {l} (N={type_counts.get(l, 0)})"
for l in labels if l in type_counts
]
g.ax_joint.legend(handles=handles, labels=new_labels,
loc="upper left", fontsize=9)
return g
# ----------------------------------------------------------
# 4. 内嵌箱线图:残差分布 + 显著性标注
# ----------------------------------------------------------
def add_inset_boxplot(joint_grid, df):
"""
嵌入水平箱线图,展示三类样本的对数残差分布,并添加显著性星号。
"""
ax_ins = inset_axes(
joint_grid.ax_joint,
width="40%", height="22%",
loc="lower right",
bbox_to_anchor=(-0.03, 0.05, 1, 1),
bbox_transform=joint_grid.ax_joint.transAxes
)
# 箱线图颜色顺序(与主图图例对应)
box_colors = [
CONFIG["colors"][2], # WGS -> 灰
CONFIG["colors"][1], # MAG -> 粉
CONFIG["colors"][0] # SAG -> 蓝
]
# ----- 专用于 sns.boxplot 的参数(含 hue + legend=False)-----
box_plot_kws = {
"data": df,
"x": "residuals_log",
"y": "type",
"hue": "type", # 新增,消除 FutureWarning
"orient": "h",
"order": ["WGS", "MAG", "SAG"],
"palette": box_colors,
"linewidth": 1,
"fliersize": 1.5,
"legend": False # 避免自动生成多余图例
}
# 绘制箱线图
sns.boxplot(**box_plot_kws, ax=ax_ins)
# ----- 专用于 Annotator 的参数(去除 legend 等无关项)-----
annotator_kws = {
"data": df,
"x": "residuals_log",
"y": "type",
"hue": "type", # 保持一致,使颜色对应
"orient": "h",
"order": ["WGS", "MAG", "SAG"],
"palette": box_colors # Annotator 也会使用该配色
}
pairs = [("WGS", "MAG"), ("WGS", "SAG"), ("MAG", "SAG")]
annotator = Annotator(ax_ins, pairs, **annotator_kws)
annotator.configure(test="Mann-Whitney", text_format="star",
loc="inside", verbose=False)
annotator.apply_and_annotate()
# 后续样式设置(不变)
ax_ins.set(xlabel=None, ylabel=None, yticks=[])
ax_ins.set_title("Residuals (log-scale)",
fontsize=9, fontweight="bold", pad=12)
for spine in ax_ins.spines.values():
spine.set_visible(False)
rect = FancyBboxPatch(
(0, 0), 1, 1, transform=ax_ins.transAxes,
facecolor="white", edgecolor="#bdc3c7", alpha=0.8,
boxstyle="round,pad=0.1,rounding_size=0.05",
clip_on=False, zorder=0
)
ax_ins.add_patch(rect)
# ----------------------------------------------------------
# 5. 主程序:执行完整分析流程
# ----------------------------------------------------------
if __name__ == "__main__":
# 准备数据
df, popt = prepare_data(CONFIG["input_file"])
# 绘制联合分布主图
g = plot_main_joint(df, popt)
# 添加内嵌残差箱线图
add_inset_boxplot(g, df)
# 自动调整布局,防止标签重叠
plt.tight_layout()
# 保存为 PDF 文件(矢量格式,适合出版)
output_file = "genome_size_vs_cds.pdf"
plt.savefig(output_file, dpi=300, bbox_inches="tight")
print(f"图像已保存至:{output_file}")
# 屏幕显示
plt.show()结语
本文展示了一套完整的基因组大小与CDS数量关系的分析流程,重点在于:
- 对数空间的线性建模与残差分析;
- 联合分布图与内嵌子图的巧妙组合;
- 统计显著性的自动标注。
这种方法不仅适用于基因组数据,还可推广到任何需要展示"全局趋势+组间差异"的场景。希望这段代码能帮助你高效产出高质量的科研图表。