论文总结
该论文比较了基于基因簇(gene clusters)的微生物组数据表示法与传统的分类学和功能谱在宿主表型分类(疾病 vs. 健康)中的表现。研究使用五个疾病数据集(2型糖尿病、肥胖、肝硬化、结直肠癌、炎症性肠病)和多种机器学习算法,发现基因簇方法在某些疾病中优于或相当于传统方法。进一步结合功能数据库(如COG、CAZy)或假设驱动的基因子集(如肠-脑轴相关基因)可提升分类性能与模型可解释性。结论:微生物组数据表示方法应因疾病而异,无统一最优方案。
摘要
随着微生物组和机器学习领域的共同进步,肠道微生物组已成为非常有兴趣的潜在生物标志物,用于宿主健康状况的分类。来源于人类微生物组的鸟枪宏基因组学数据是由一组高维的微生物特征组成的。使用这些复杂的数据来建模宿主-微生物组的相互作用仍然是一个挑战,因为保留从头开始的内容会产生一个高度粒化的微生物特征集。在本研究中,我们根据鸟枪法宏基因组学中不同类型的数据表示,比较了机器学习方法的预测性能。 这些表示方法包括常用的分类学和功能谱以及更细粒度的基因簇方法。对于本研究中使用的5个病例-对照数据集( 2型糖尿病、肥胖、肝硬化、结直肠癌、炎症性肠病),基于基因的方法,无论是单独使用还是与基于参考的数据类型相结合,都可以获得与分类学和功能轮廓相似或更高的分类性能。此外,我们表明从基因的特定功能类别中使用基因家族的子集,突出了这些功能对宿主表型的重要性。 本研究表明,无基准微生物组表征和宏基因组注释都可以为基于宏基因组数据的机器学习提供相关表征。
在使用宏基因组数据时,数据表示是机器学习性能的重要组成部分。在这项工作中,我们表明不同的微生物组表示提供了不同的宿主表型分类性能,这取决于数据集。在分类任务中,非靶向的微生物组基因含量可以提供与分类学分析相似或改进的分类。基于生物功能的特征选择也提高了一些病理的分类性能。基于函数的特征选择结合可解释的机器学习算法可以产生新的假设,这些假设可以潜在地进行机制分析。 因此,这项工作提出了用于机器学习的微生物组数据表示的新方法,可以增强与宏基因组数据相关的发现。
引言
近年来,微生物组特征与人类疾病之间的联系已被广泛报道,特别是在慢性疾病中( 1-5 )。许多研究已经将机器学习( ML )应用于广泛的微生物组数据集,以收集隐藏的知识,更好地理解肠道微生物组在健康和疾病中的应用( 6-11 )。在推论的语境中,在用于疾病分类的宿主表型中,这种建模可以用于指向重要的微生物生物标志物或作为潜在的诊断工具。高通量测序技术允许对微生物组进行深度表征,包括不可培养微生物( 12 )。然而,需要对宏基因组数据进行处理,以允许它们与宿主表型之间的关联。鸟枪法基因组学研究常使用基于参考的数据类型,如分类学和功能谱( 6 )。例如,MetaPhlAn软件从宏基因组数据中计算分类学组成,其结果类似于16S宏分类学分析( 13 )。相反,HUMAnN提供了已知代谢途径的宏基因组的功能注释( 13 )。 构建微生物群落功能谱的最大挑战可能是将基因与特定功能联系起来的困难。的确,39 .在人类肠道微生物组的综合集成基因目录( Integrated Gene Catalog,IGC )中,有6 %未映射到功能数据库,其余15 - 20 %以前已经观察过但没有已知功能( 14 )。使用宏基因组de novo组装可以帮助克服这些限制,因为组装中编码的信息不限于数据库的内容。一些机器学习算法已被用于从微生物组数据中分类宿主表型,从随机森林( RF )到基于深度学习的方法( 15、16 )。 已经建立了数据库,以简化用于机器学习目的的微生物组数据的访问( 17 )。大多数研究使用分类学轮廓作为预测的输入数据,或基于16S代谢组学数据,或基于鸟枪数据( 18-20 )的分类学轮廓。利用公开的16S代谢组学数据,吉利贝蒂和合作者观察到,基于微生物组数据的宿主表型分类主要由微生物类群的存在与否驱动( 21 )。将微生物组分析与代谢组学( 15 )或宿主相关变量( 22 )相结合的多组学方法也越来越多地被发表。 Lee和Rho利用深度神经网络获得了一种嵌入表示,成功地将基因组水平的数据与分类学和功能轮廓相结合,提供了改进的分类( 23 )。多分类策略也被提出( 24 ),其中使用单一算法对几种疾病进行分类。受生态系统启发的机器学习方法已经显示出很有前途的结果( 25 )。这种多组学方法的一个有趣的例子是通过微生物组和临床数据预测对食物的血糖反应( 26 )。关于使用宏基因组基因含量进行机器学习分类的现有文献仍然很少,尽管它确实表明这种方法是一种很有前途的途径。 事实上,Le Goallec和合作者的工作表明,使用基因含量进行表型预测可能优于分类学分析( 7 )。机器学习也被用于从卵子NOG或KEGG数据库注释的微生物组基因中识别2型糖尿病( Type 2 diabetes,T2D )的生物标志物( 27 )。在这项工作中,我们比较了基于shotgun宏基因组学的不同数据表示策略的疾病分类性能。我们的假设是,与分类学描述相比,基于基因的颗粒微生物组表示可以提供更好的分类和可解释性。除了总的微生物组基因含量外,我们还使用了基于现有数据库的基因家族的curated子集,以及基于分子功能的假说驱动选择。 我们从具有公开可用原始数据的病例对照研究中选取了5个鸟枪宏基因组学的人类微生物组数据集。本研究纳入的5个数据集分别来自2型糖尿病( T2D , n = 199) ( 2篇)、肥胖( OB , n = 265) ( 1篇)、肝硬化( LC , n = 237) ( 4篇)、结直肠癌( CRC , n = 141) ( 5篇)和炎症性肠病( IBD , n = 124)研究( 3篇)。对于每个数据集,我们在一个由健康和患病个体的二分类组成的预测任务中评估数据表示的效果。 我们研究中使用的数据表示是分类谱( MetaPhlAn3 ) ( 13 ),潜在的代谢功能谱( HUMAnN3 ) ( 13 ),70 %一致性的基因簇,以及基于参考数据库或假说驱动蛋白结构域选择的基因簇的不同子集。对于每种数据类型,我们评估了9个分类器的预测性能:两个支持向量机( SVM )。线性核函数( L1 -和L2范式),具有径向基核函数的SVM ( SVMrbf ),两个逻辑回归( LRs ) ( L1 -和L2范式),决策树( DT ),随机森林( RF ),集合覆盖机( SCM ) ( 28 )和随机集覆盖机( rSCM ) ( 29 ),由SCM衍生的集成算法。对于每个数据集,我们在机器学习协议之前应用一个特征选择随机森林。我们使用不同的指标对最终的模型进行了评估,特别是对不平衡类的平衡精度,以及受试者工作特征曲线下的流行区域( roc AUC )。图1总结了我们的实验方案。
结果
基因含量是微生物组分类和功能谱的替代指标
我们的第一个目标是确定在从鸟枪法宏基因组学数据中分类健康/疾病的机器学习问题中,基因簇丰度是否比分类学丰度表现更好、相似或更差。为了做到这一点,我们比较了每个数据集的数据表示和使用基于样本分裂的具有随机个体效应的广义线性模型( GLM )的机器学习算法的性能。我们比较了9种不同机器学习算法(图2为最佳算法,附图S1至S5为所有算法)利用基因簇和分类学相对丰度获得的平衡精度。 微生物组表征的性能一般取决于用于机器学习的算法,表现最好的组合通常在不同数据集之间存在差异。F1得分和AUC结果分别见附图S6 ~ S15。当考虑每种表示的最佳执行算法时,基因簇的平衡准确率显著高于OB的分类法,但并不显著。与T2D,IBD和CRC的分类不同,并显著低于LC的分类(图2 )。这些结果表明,基因簇以类似于分类学的方式编码疾病预测的相关信息,尽管对于某些问题/算法组合,一种表示优于另一种表示。我们还研究了基因簇和分类学如何与使用HuMANN3软件(识别为图2中的函数)确定的微生物组功能图谱进行比较。仅在OB中,基因簇平衡精度显著高于HuMANN3功能。分类学仅在LC中显著高于HuMANN3功能。使用基因簇、分类学和功能的组合进行学习(确定为图中的Fusion ) 2 )仅在OB中显示出显著高于分类学的信号。尽管基因簇的维数很高,但使用初始随机森林选择的特征通常可以在5个数据集中获得与分类学丰度或功能图谱相当的分类性能。
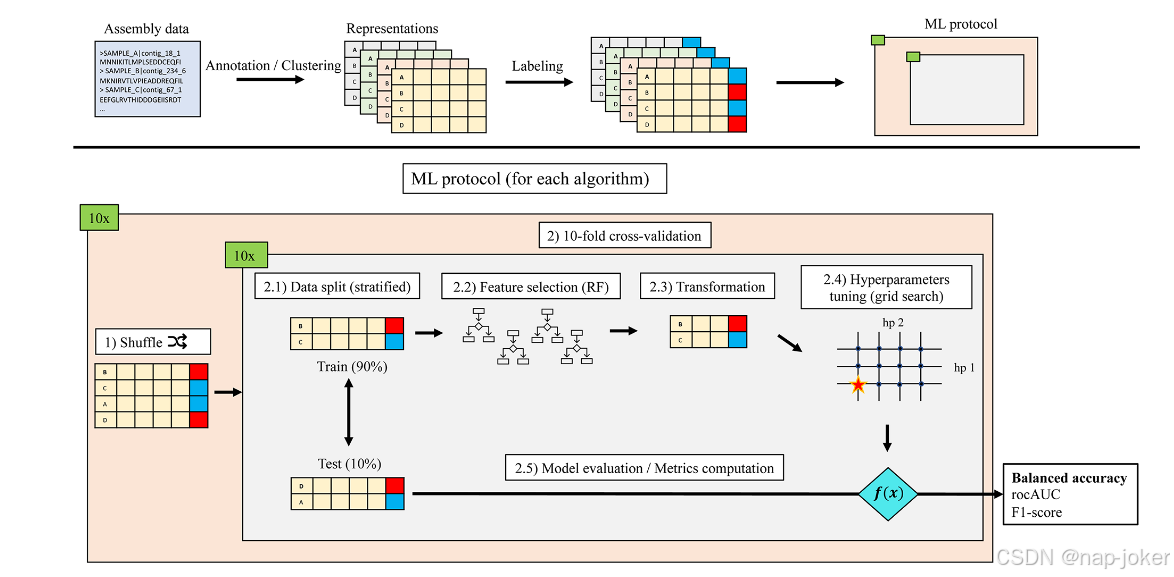
图1生物信息学和机器学习协议示意图。每个表示作为机器学习( ML )协议的输入。( 1 ) 10折交叉验证的每10个重复用一个不同的随机种子启动。为了限制随机化偏差,每个拆分中的例子对所有的表示都是相同的。( 2 . 1 )对于每一个分割,将训练集和测试集按照类别比例进行分层。( 2 )采用随机森林( Random Forest,RF )进行重要特征的选择。( 2.3 )只保留选定的特征进行下一步操作。( 2.4 )针对每种算法,对一组超参数( hps )进行了全面的测试,并保持了最佳平衡精度的组合。 计算其他指标:受试者工作特征曲线下面积( roc AUC )和F1值。
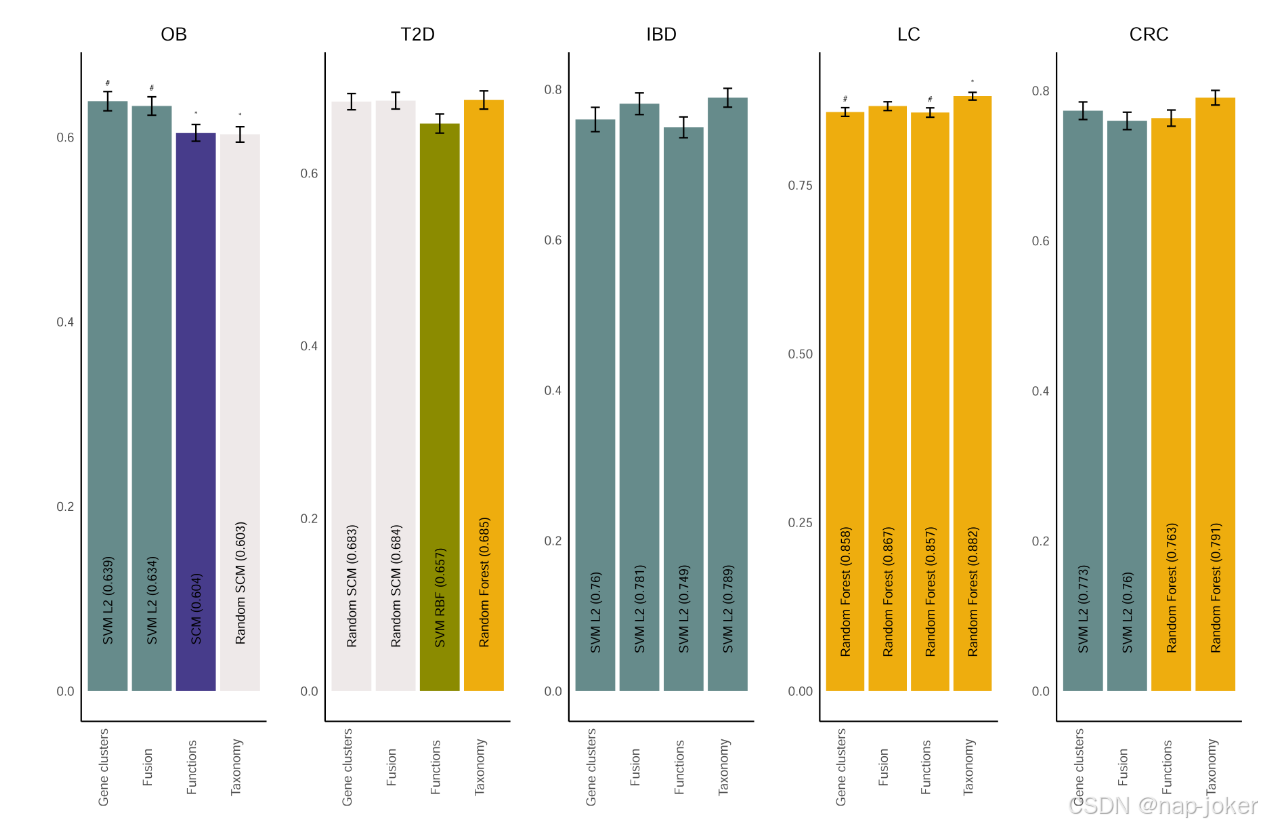
图2宏基因组数据表示方法以数据集依赖的方式影响机器学习分类结果。比较结直肠癌( CRC )、炎症性肠病( IBD )、肝硬化( LC )、肥胖( OB )和2型糖尿病( T2D )的基因簇、基因功能、分类学和三种表征的融合在病例/对照分类中的表现。采用平衡准确性对性能进行评估。对于每种数据表示,表现最好的算法的平均平衡精度用表示95 %置信区间的误差条表示。每个表示的最佳执行算法的名称在条形图中显示。 条形图上的符号表明,经过错误发现率( false discovery rate,FDR )校正后的( P < 0 . 05 ))与分类学( # )或基因簇( * )相比,数据表示的平衡精度有显著差异。
COG分类有利于表型预测的可解释性
我们研究了基于COG数据库选择的基因簇的分类潜力,该数据库允许对26个广泛功能分类中的细菌基因进行分类(图3为最佳算法,所有算法的附图S16 ~ S20,附图S21 ~ S25为F1得分,附图S26 ~ S30 )。for AUC )。根据先前的实验,我们假设,对于特定的疾病,某些COG类别可以改善疾病表型预测,因为特定的基因功能可能与表型相关。此外,使用较小的特征集可以显著减少模型构建所需的资源。因此,我们根据序列相似性将基因簇分配到COG类别中,并对每个新的基因簇子集使用与之前相同的ML协议。对于结直肠癌和肝硬化,通过COG类别过滤的基因簇获得的平衡准确率显著低于分类学。 相比之下,对于肥胖表型预测,两个COGs具有显著高于分类学的平衡准确性,即氨基酸转运和代谢( E )和碳水化合物转运和代谢( G )。在IBD表型预测中,辅酶转运和代谢( H )提供了最高的平衡准确性,尽管这种差异与基因簇和分类学相比没有统计学意义。最后,对于2型糖尿病,COG类别细胞运动性( N )比分类学和所有基因簇具有更高的平衡准确性,但不具有统计学意义。该模型使用SCM算法生成,该算法提供了稀疏的可解释模型。 在本例中,来自罗斯氏菌和毛螺菌属的鞭毛蛋白输出ATP酶Fli I均属于毛螺菌属科,与健康状态相关。As是来自Phascolarctobacterium faecium的prepilin - type N-terminal cleavage / methylation domain-containing protein。总的来说,这些结果表明,在某些情况下,基因簇的功能分类可以提供比普通方法更好的预测,此外还有利于可解释性。
基于数据库的特征选择
为了研究基于基因特定生物学特性的特征选择的使用,我们测试了基于特定生物学功能选择的特定组基因簇的方案。每个类群都包含至少一个序列与已知功能基因相似的基因簇。 我们使用的数据库来自特定功能类别的基因:涉及使用碳水化合物的酶碳水化合物-活性酶( CAZy )数据库( 30 ),抗生素抗性基因 Mobile Elements and Resistance Genes Enhanced for环境基因组学( MERGEM )数据库( 31 ),插入序列( MERGEM ),生物合成基因簇关于生物合成基因簇的最小信息( MIBiG )数据库( 32 )和来自综合酶数据库BRENDA ( 33 )的酶。对于5个数据集,基于基因簇子集的表示并没有显著优于使用所有基因簇的表示(图3 ) 图4为最佳算法,所有算法的补充图S31 ~ S35,F1得分的补充图S36 ~ S40,AUC的补充图S41 ~ S45 )。然而,在IBD数据集中,来自MERGEM数据库的插入序列和抗性基因显示出比基因簇和分类学更高的平衡精度,尽管差异不具有统计学意义。虽然基因簇的某些子集的性能通常不优于基因簇或基于分类学的预测,但我们认为领域知识是一种潜在的工具,可用于使用宏基因组基因含量预测表型的特征选择。
面向项目特定可解释性的假说驱动特征选择
作为概念验证,我们设计了一个基于特定假说相关分子功能的基因簇选择实验。在这方面,我们假设含有Pfam(34)结构域的微生物基因,这些结构域来自与内源性大麻素类似物产生和降解相关的酶,或潜在的肠道脑轴效应因子,可能编码肠道微生物组与代谢疾病之间交叉调音的相关信息(35--37)。有趣的是,含有肠脑轴(GBA)相关结构域的基因相比其他表示,在预测肥胖方面实现了最高的平衡准确率(见图4,即GBA)。事实上,GBA基因子集在OB数据集,且准确率比基因簇更高,但这一差异在统计学上并不显著。该模型采用L2正则化数据和逻辑回归算法获得。由于用于构建该模型的基因簇子集与特定假说相关联,我们调查了生成模型中重要的特征,并结合现有知识进行了解释。因此,我们对所有生成的100个模型的系数求和至少一个模型中包含的特征,汇总了用于预测的特征列表。我们调查了系数总和大于0.5或低于−0.5的基因簇(见图5)。该模型中,14个基因簇的数值为>0.5,而68个的数值低于−0.5。大多数基因簇与梭菌纲中的基因相似,具体包括梭菌科、真细菌科、拉克诺螺科和虹黄色螺旋体科。拟杆菌科基因簇是肥胖的积极预测因子,而许多其他分类单元则与肥胖负相关,包括阿克曼科、双歧杆菌科和卵菌科,这些通常与良好的代谢健康相关(38, 39)。特别是,GNAT家族的N-乙酰转移酶结构域在模型中包含的基因簇中代表性最高,主要见于如前所述的拟果科(40)及若干梭状菌科。这些酶参与合成N-酰基酰胺,包括脂胺酸和内源性大麻素类似物,它们是G蛋白偶联受体和核受体(即过氧化物酶体增殖激活)受体的激动剂,这些受体在生理和病理代谢复苏中发挥重要作用(40)。来自Clostridiaceae的β内酰胺酶Superfam观察到最强的负系数。在人类中,金属β内酰胺酶可能参与生物活性脂质代谢,例如NAPE-PLD酶,它催化内源性大麻素、阿南酰胺及其他N-酰基乙醇胺的生物合成(41)。这些结构域在Clostridia科和物种中的差异分布可能解释了这些分类单元在代谢控制上的双重性质(42)。这一概念验证实验展示了机器学习中的假设驱动数据表示。此外,这种方法使我们能够从新颖的视角研究一个经过充分研究的数据集,并提供可能带来新发现的洞见。
讨论
本研究探讨了散弹枪式微生物组数据的不同表现如何影响五项分类任务中健康和疾病表型分类的能力。我们观察到,所有病理状况中表现最佳的代表性并不相同,这表明目前尚无一套放之而皆准的数据表示方法来预测肠道微生物组数据中的疾病。事实上,数据表示和机器学习算法表现最佳的组合在五个问题中各不相同,每个问题由一个研究代表。微生物组数据中最常用的表示方式,即分类丰度,在此例中是利用MetaPhlAn3确定的,其准确性始终高于所有针对结直肠癌和肝硬化的随机森林算法测试的其他表示。我们精心整理的可能参与与肠-脑轴相关分子生成的基因集合,利用L2正则化逻辑回归为肥胖提供了最佳的预测特性。对于T2D,与细胞运动性相关的基因(COG类别N)通过SCM算法提供了最佳预测。最后,使用L2正则SVM算法,与移动遗传元件相关的基因为IBD提供了最佳预测。因此,我们的主要结论是应比较不同的微生物组表征,以基于散弹枪式微生物组数据提升表型分类。我们最初假设,来自散弹枪式微生物组数据的完整基因内容相较于分类组,能够提升表型预测,而这一预测仅在产科分类问题中得到验证。基因家族有潜力编码仅使用分类组合时丢失的信息。由于功能未知的基因不会从数据中被消除,这些基因中可能编码的重要生物学信息可能会提高预测的质量。基因簇可能包括代表物种甚至菌株的基因。它们甚至可能解释可以在不同物种或基因家族之间进行地平线计数转移的基因,这些基因可能被无关的分类单元共享。为了提升机器学习模型的可解释性并降低数据集的初始特征大小,我们对基因家族进行了与基因数据库或COG类别的相似性过滤。这种方法使我们能够筛查特定功能与疾病的关联,同时保持特征的细粒度。类似的过滤方法此前也被用于比较微生物基因组的k-mer含量,以确定特定基因功能是否可能与物种内亚群相关(43)。在这里,我们观察到使用特定基因子组训练机器学习模型时,性能优于分类法,表明该方法在宏基因组数据的机器学习中具有潜在应用价值。此外,在这些较小数据集上学习模型所需的计算时间和内存远低于使用所有基因簇,但性能相当甚至更优。T2D数据集中观察到一个显著的潜力例子,稀疏的SCM算法能够精准定位与细胞运动相关的基因。来自鞭毛螺旋藻科的鞭毛蛋白导出ATP酶FliI和粪便形畸形杆菌的前毛蛋白型N端切割/甲基化结构域蛋白被鉴定为T2D的潜在生物标志物。事实上,Lachnospiraceae在以往研究中已被关联T2D(2, 44),但目前稀疏的机器学习模型提供了基于两个分类单元特定基因,有助于更好地理解该疾病的机制。因此,这种方法有潜力揭示与疾病表型相关的基因。基因级数据类型在宿主表型预测中的预测表现可能表明,对于某些疾病,微生物功能与表型的关系比微生物本身更为密切。此外,数量稀疏针对特定基因的模型更适合跨队列验证和潜在临床应用(6)。我们还基于选定的Pfam结构域进行定制基因家族选择,研究疾病与可能参与肠道-脑轴相关代谢物合成或降解的基因之间的潜在关联。肠道原核生物可能合成神经活性介质的潜力,此前已有研究表明,例如多巴胺相关代谢产物(45)。最近,共生肠道细菌还被证明能产生内源性大麻素类似物,这些类似物有可能与宿主的GPCR信号系统相互作用,调节肠道和代谢功能(40)。基于此,我们假设与内源性大麻素样分子产生和降解相关的酶域,如N-酰基β-泰德氨基酸和N-乙基乙醇胺,以及神经递质,可以编码关于菌群失调、扩展内源性大麻素系统与疾病之间交互作用的新信息。这也可能凸显微生物肠道-大脑轴效应器在精神和神经系统疾病以外的其他情境中潜在作用。例如,内源性大麻素同源和类似物已被证明参与代谢疾病、肠道过程和精神障碍(46, 47),因为它们可以作为激动剂或受体拮抗剂,作用于这些系统中不同的受体。微生物群可以生物合成多种代谢物,每种氨基酸-脂肪酸组合以酰胺形式存在,可能对宿主代谢或心理表型产生正负影响,类似于某些宿主产生的N-酰基氨基酸(48--50)。作为肥胖预测因子的GNAT N-乙酰转移酶同源物也可能参与此类过程(40,51--54)。尽管这些结果仍属初步阶段,但它们展示了以假设为驱动的方法在微生物组特征选择中的巨大潜力,有望启发未来的机器学习方法,并为现有数据集的研究提供新视角。使用基因家族进行表型预测的一个挑战是矩阵大小,这需要大量内存和计算来生成模型。例如,T2D任务(n=199)的机器学习协议在使用基因簇矩阵时占用6:09:01(h:m:s)和65.5GB内存。筛选基因簇产生的数据集明显更小,因此资源需求更小。对于集群匹配插入序列(MERGEM_IS),协议占用1:58:38和1.16 GB内存。相比之下,分类学和功能配置文件(MetaPhlAn3和HUMAnN3)分别为2:48:06(0.223 GB)和06:13:48(0.300 GB)内存。关于其他数据类型所需资源的信息可见补充表S1。高维数据类型的另一个风险是过拟合,即学习的是主体特定的特征而非类别定义特征。为解决此问题,我们采用了一种嵌入式特征选择技术,即训练随机森林,根据所得模型对特征进行排序,并对这些排序特征的子集进行学习,这一方法此前已有使用(6, 11)。将共丰基因组聚类也可能有助于高效地减少特征(55)。未来,应研究其他数据简化方法,以支持使用粒度宏基因组数据,如基因内容或k-mers(56),这些数据既能实现核苷酸解析,又能产生具有数十亿特征的巨大特征大小。该分析的另一个潜在挑战是存在众多相关特征。即使在分类学层面,测量成对相关性在计算上也具有挑战性。由于一个包含n个特征的数据集中可能的两特征相互作用数量为n × (n − 1)/2,这通常导致数百万次计算,因为微生物组由数千个微生物组成。此外,由于微生物生活在一个群落中,很可能存在三特征、四个特征或更多特征的相互作用(57)。当使用基因层面的微生物组数据时,这些问题会加剧,因为这些基因可能由相关微生物携带。预测模型中使用的基因可能是特定分类群的代理,而非直接关联预测表型的功能。这些元素应该在基因级宏基因组数据的降维技术开发中被考虑。本研究的一个局限是每个分类问题只能使用单一队列。这限制了结果的传播范围,因为数据表示和学习算法的最佳组合可能无法迁移到使用其他数据集的类似分类问题上。此外,本研究中解释的模型可能无法迁移到其他数据集。因此,这些理论应被视为未来作品中需要验证的假说。无论哪种情况,模型解释后应结合外部队列进行实验验证。尽管如此,利用这五个文献中研究充分的分类问题,我们能够利用宏基因组数据证明基因簇或基因簇子集在分类中的潜在效用。
总结
总之,我们的结果表明,在基于微生物组数据的机器学习预测疾病状况时,应对不同的数据表示进行分析。我们证明,通过分类组成和基因簇可以获得类似的预测。根据公开数据库或与科学假说相关的人工整理属性选择基因子集,可以提高预测某些病理的准确性和可解释性。在使用基于粒度特征(如基因簇)的表示时,应考虑产生稀疏模型的算法,因为它们提供了生物标志物,这些标志物有可能移植到独立的验证队列,甚至在体外进行测试。
材料与方法
不同数据类型的生成
MetaPhlAn3和HUMAnN3分别生成的分类和功能特征(13)通过R软件包CuatedMetagenomicData(版本1.20.0)(58)下载。IBD和T2D数据集分别以ERA000116号和PRJNA422434号从NCBI序列读取档案下载。OB、CRC和LC数据集分别从欧洲核苷酸档案(ENA)下载,入藏编号分别为PRJEB4336、PRJEB6070和PRJEB6337。基因聚类矩阵的生成过程有三个:利用MEGAHIT(版本1.2.9)(59)将原始读段组装成contigs,预测蛋白质编码基因并随后使用Prodigal(默认参数)将其翻译成氨基酸(版本2.6.3)(60),以及使用CD-HIT在70%同一阈值处聚类所得氨基酸序列(版本4.7)(61),参数为-c 0.70 -aS 0.90 -d -M 0 -T 0 -g 1 -G 0。为了衡量聚类参数对预测表现的影响,我们分别评估了{0.70, 0.85, 0.95}和{0.70, 0.90}集合中的身份阈值c和覆盖阈值aS。c和aS的0.70和0.90的整体结果略高于其他组合(补充图S46),因此被选用于后续步骤。融合数据类型由三个表格的连接生成:分类和功能谱以及基因簇。为了制作基因簇的筛选表,我们首先将数据库中的基因编码序列和样本的基因编码序列合并。本次分析所用序列来自CAZy(30)、COG(62)、MERGEM(31)、BRENDA(33)和MIBiG(32)数据库。接下来,我们保留了数据库中至少包含一条序列和样本中一条序列的基因簇。为选择可能参与神经递质及内源性大麻素类似物产生或降解的基因簇,我们构建了一个本地数据库,收集了来自Pfam的蛋白质结构域的隐马尔可夫模型(HMM)剖面,这些结构域出现在神经递质、内源性大麻素及内源性大麻素类似物合成和降解途径中的酶中(补充表S2)。随后,我们在每个基因序列中搜索了这些HMM谱该簇的代表序列,使用HMMER(63)命令hmmsearch(-E 0.001)并保留所有代表序列至少匹配一个HMM谱的基因簇。对于所有数据类型,仅存在于一个样本中的特征被剔除,因为它们被认为无信息。
机器学习协议
该机器学习协议是在 Python(版本 3.6.3)中开发的,使用 scikitlearn(版本 0.22.1)。SCM (28) (https://github.com/aldro61/pyscm) 和 rSCM (29) (https://github.com/thibgo/randomscm) 算法通过他们的 Github 仓库下载。分类表现通过10次交叉验证评估,重复10次独立运行,得出100种不同的数据分割。对于每个拆分,训练集(90%的示例)和测试集(10%的样本)在特征选择前被分离。因此,在对基于训练集构建的模型进行最终评估之前,算法并未看到该测试集。协议的第一步是在预处理步骤后的所有剩余特征上应用一个简单的RF------scikit-learn的RandomForestClassifier,包含100个决策树(n_estimators)及所有其他超参数默认设置。特征按照"features_impor tances_"方法排序,该方法基于基尼杂质得出的评分,得分越高,特征的重要性越高。选择重要性大于重要性均值的特征用于最终模型的训练。对每个算法,测试一组超参数,并保留得分最高的组合。为了考虑每个分类任务的不平衡类别,我们用平衡准确率评分评估模型表现。模型使用广义线性模型(GLM)与 NLME R 软件包(版本 3.1--148)比较了每个数据集。平衡准确度通过拟合于LME的排名值(~数据表示*算法)进行归一化,显著性则通过方差分析(ANOVA)检验,随机效应嵌套于样本分割中。具体比较使用Contrast R软件包(版本0.22)进行评估。多项检测中采用假发现率<0.05校正的P值被视为显著
算法与超参数
SVM是一种旨在找到最大化各类样本间距的超平面的算法(64)。当类不可线性分离时,可以用核函数将数据映射到更高维空间,从而找到超平面。对于超参数调优,正则化参数C被选入集合{10⁻⁻, 10⁻³, 10⁻², 10⁻¹, 0.25, 0.5, 0.8, 0.9, 1, 1, 10, 100, 500, 1,000},适用于带有线性核和rbf核的SVM。在L1和L2正则化LR中,C参数也使用相同的值。对于DT,树的最大深度为{1, 3, 5, 10, 25}。分割节点所需的最小样本数为{2, 5, 10},叶节点所需最小样本数为{1, 2, 4}。对于RF,树的数量固定为500棵。分开节点所需的最小样本数、树的最大深度以及叶节点所需的最小样本数分别位于{2, 5, 10}、{1, 3, 5, 10, 25}和{1, 2, 4}。SCM是一种基于规则的算法,学习合取(逻辑与)和析取(逻辑或),这两者是规则的逻辑组合(28,56)。对于SCM,效用函数的权衡参数为{0.5, 1, 2},最大规则数为{1, 2, 3, 4, 5},模型类型在合取和析取之间变化。rSCM 是一种集合算法,使用带有引导法聚合的 SCM。对于rSCM,基估计量的数量为{30, 100},训练每个估计器的样本和特征比例均为{0.6, 0.85},效用函数的权衡参数p为{0.1, 0.316, 0.45, 0.562, 0.65, 0.85, 1.0, 2.5, 4.39, 5.623, 7.623, 10.0}。