
1. 小编导读
在微生物组研究中,我们常常受困于一个两难选择:既希望像UniFrac那样利用序列间的亲缘关系来评估群落差异,又头疼于系统发育树构建过程的高昂计算开销和某些基因区域(如真菌ITS)几无可靠的系统发育信号。Bokulich在2025年发表的这篇方法学论文,将k-mer频率计数正式引入了微生物多样性分析的流程。所谓k-mer,就是一条序列中所有长度为k的子串。研究表明,基于k-mer频率的beta多样性距离,与加权UniFrac距离的相关性高达0.795(Mantel检验),并且在真菌全长ITS序列的分析中,k-mer方法能明显提升样本类型的区分度 。更诱人的是,k-mer计数比传统的"mafft比对+FastTree建树"流程快一到三个数量级。
2. 摘要
k-mer频率信息在生物学序列分析中应用广泛,涵盖物种分类、序列相似度估计和监督学习等领域。然而,尽管其用途多样,k-mer计数在多样性评估方面却长期被忽视。研究系统分析了k-mer计数在微生物标记基因测序数据(16S rRNA基因及真菌全长ITS序列)的alpha多样性、beta多样性以及监督分类中的应用潜力。结果显示,基于k-mer频率的多样性度量与基于系统发育的多样性指标(如UniFrac和Faith's PD)具有高度对应性,且在某些方面表现出明显优势,尤其是在缺乏可靠系统发育信息的场景下(例如ITS区域)。k-mer计数可在无需进行昂贵的两两序列比对的前提下,将序列的亚序列组成信息整合进多样性评估。作者将其定位为一种对传统方法的补充手段,能够支撑大规模、无参考数据库依赖的微生物群落分析,适用于生物地理学、生态学及生物医学数据。相关方法已实现为QIIME 2插件q2-kmerizer (https://github.com/bokulich-lab/q2-kmerizer)。
3. 研究方法
k-mer计数核心流程
方法学本身并不复杂。程序首先读入所有样本中观察到的ASV的序列,利用scikit-learn中的CountVectorizer(或可选的TfidfVectorizer)将每条序列视为一个"文档",并将字符形式的k-mer作为"词条"进行枚举。随后,通过NumPy的矩阵乘法,将每个样本中ASV的丰度矩阵乘以各ASV所含k-mer的组成矩阵,从而得到每个样本的k-mer频率特征表。若启用TF-IDF加权,则能够上调那些具有高区分度、较少共享的k-mer的权重,同时抑制普遍出现在各类序列中的低信息量k-mer。整个流程中,用户可通过min_df(最小出现频次)过滤稀有的k-mer,通过max_features限定所使用的最大k-mer特征数目。
基准测试数据
研究分别在EMP和GSM两个数据集上进行了验证。原核生物部分使用地球微生物组计划(EMP)的16S rRNA V4区数据,经去噪获得ASV,过滤掉总观测数少于10次的低频特征后,将所有样本均匀抽平至5000条序列,最终得到975个样本。真核生物部分使用了全球土壤真菌组联盟(GSM)的PacBio全长ITS数据,经预处理保留300--800 nt的序列,以100例为一组,从七个最丰富的生物群落中随机抽样组成678个样本的测试集,并抽平至3000条序列。在参数对比中,为与ASV数对齐,EMP数据的max_features设为5000,GSM数据的max_features设为10000。
多样性与监督学习评估
特征计算完毕后,即可使用QIIME 2的标准多样性插件执行alpha与beta多样性分析。beta多样性调用了Bray-Curtis、Jaccard、Aitchison距离等非系统发育指标,并通过Mantel检验与加权/非加权UniFrac距离进行相关性对比。PERMANOVA用于检验样本分类(如EMPO 3环境类别)的组间分化效果。监督学习方面,采用十倍嵌套交叉验证的随机森林(500棵树),分别以ASV频率、k-mer频率以及基于UniFrac距离的k近邻分类进行样本类别预测,并使用ROC曲线下面积(AUC)衡量性能。所有测试均在单核环境下进行,记录了不同序列数量与长度下的运行时间。
参数优选策略
k-mer长度的选择对结果有直接影响。为避免"数据窥探"式的p值操纵,作者仅依据主坐标分析(PCoA)的聚类质量和轮廓系数(Silhouette score)等定性指标来确定最佳k值,最终选定k=16作为适用于EMP和GSM数据的通用默认值。研究建议使用者在自己的实验中也应采取类似的定性评估方案,或使用独立数据集进行参数调优。
4. 主要结果
k-mer beta多样性与系统发育指标高度一致
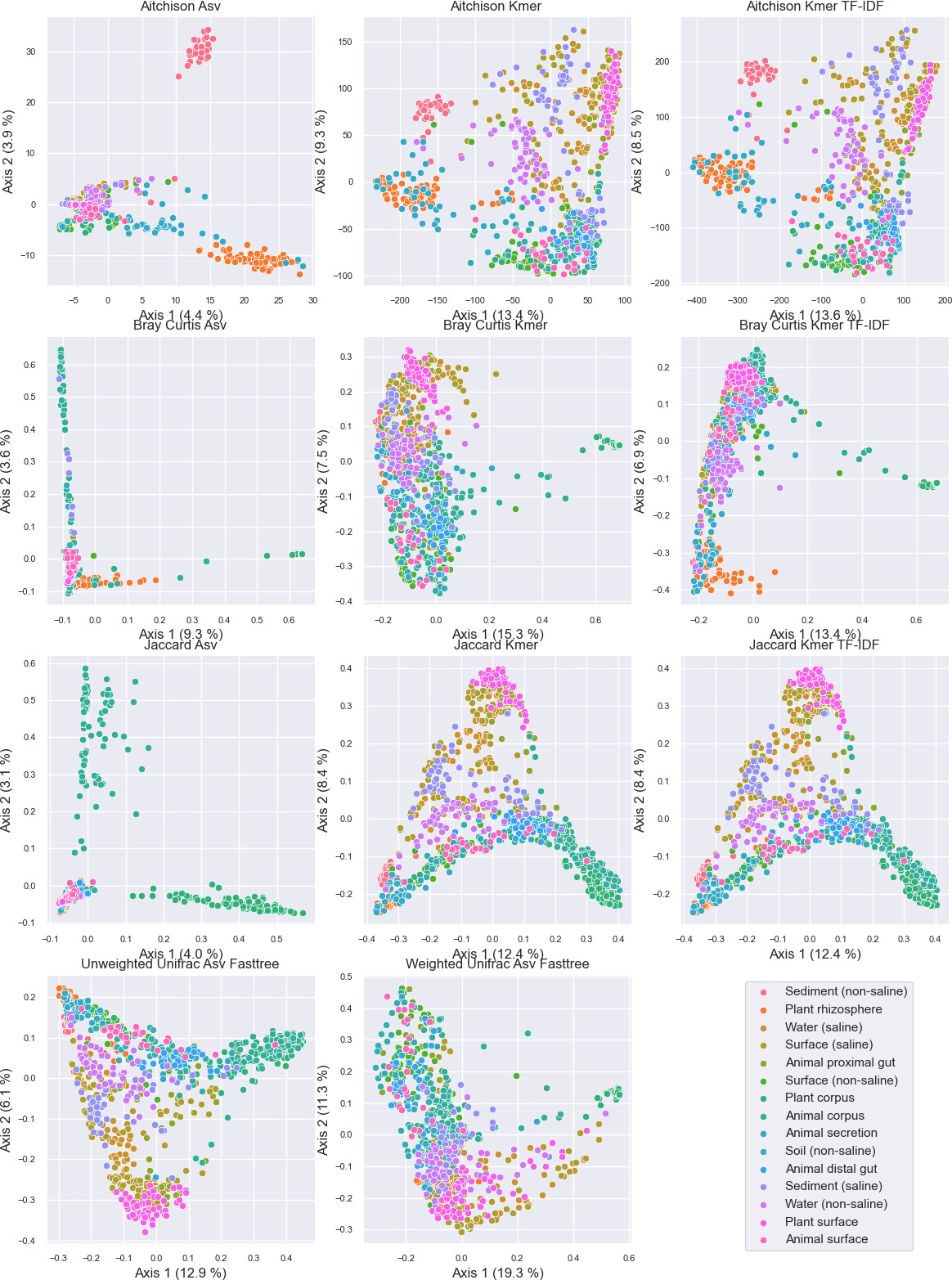
在EMP数据集上,基于16-mer频率的Bray-Curtis或Jaccard距离,与基于ASV系统发生树的加权UniFrac、非加权UniFrac距离呈现出极高的一致性。Mantel检验表明,16-mer Bray-Curtis距离与加权UniFrac的Spearman相关系数高达0.795,而传统ASV的Bray-Curtis距离与加权UniFrac的相关性仅为0.405。通过PERMANOVA定量衡量,k-mer频率距离对样本环境类别(EMPO 3)的解释方差比(R²)显著高于基于ASV的传统距离,且与UniFrac的R²值非常接近。这意味着,仅靠k-mer计数就几乎实现了系统发育加权分析的效果,能够为样本间遗传组成差异提供敏感度量。
提升监督学习的预测准确度
当使用随机森林对样本环境类别进行分类时,基于k-mer频率(无论是否进行TF-IDF加权)的模型准确度可达0.854--0.855,略优于基于ASV频率的0.828,且远超基于加权UniFrac距离的k近邻分类(准确度0.787)。这提示通过k-mer化扩展特征空间,可以在不增加太多计算负担的情况下提升下游分类任务的精度。
k-mer长度与过滤参数的影响
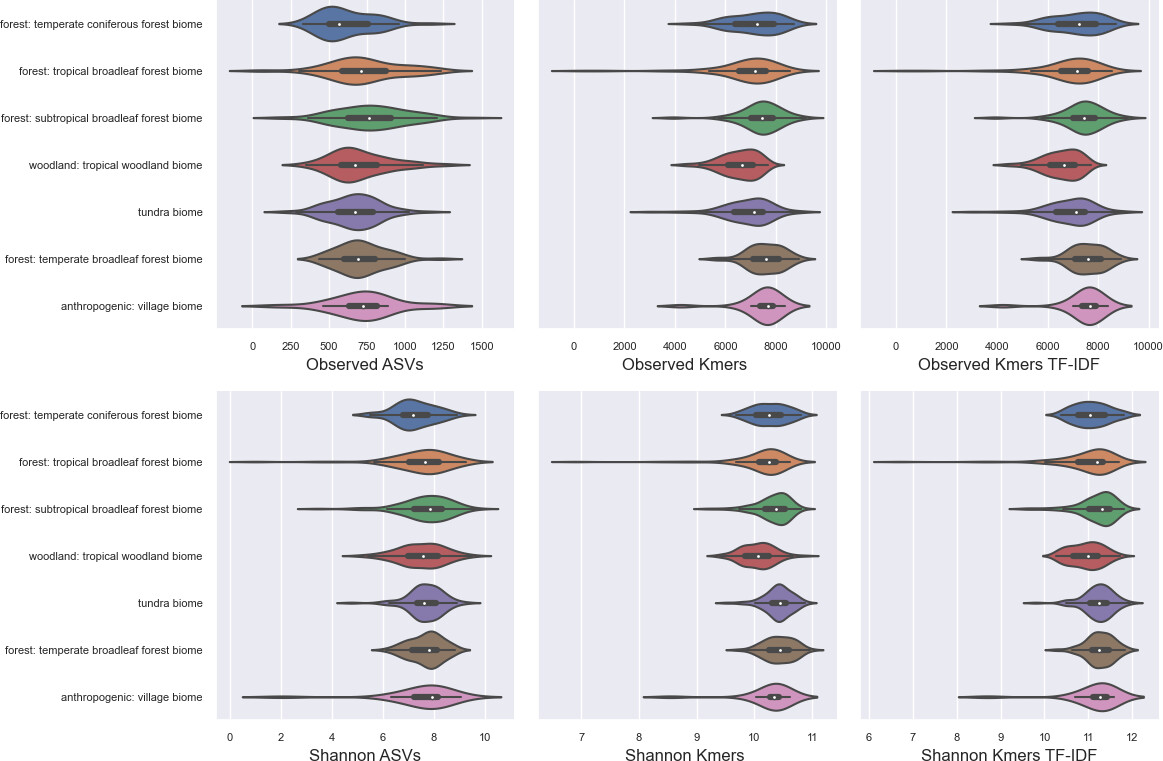
在固定最大特征数的情况下,k-mer长度过低会导致特征空间坍缩,样本区分度变差;过长的k-mer则可能过于特异,同样削弱信息。对EMP数据而言,k=7开始轮廓分数显著上升,k=12--32均能提供良好的PcoA拟合度,k=16作为折衷选择。此外,适度的采样深度抽平、以及对k-mer频次的合理过滤是保证分析稳健性的前提,但进行alpha多样性分析时,务必避免使用TF-IDF和激进的特征筛选,以免人为扭曲真实的k-mer丰富度和均匀度。
解决真菌ITS无法建树的痛点
GSM全长ITS数据的案例尤其具有说服力。基于ASV的beta多样性降维图解释方差极低(前两个主轴<5.7%),且各类生境样本交叠严重。而将同样的16-mer频率应用于Bray-Curtis、Jaccard或Aitchison距离后,前两轴解释方差飙升至29.4%--34.7%,不同生物群落的样本清晰分离。PERMANOVA的效应量平均提高了0.067--0.121。在alpha多样性方面,基于k-mer丰富度和香农熵的组间比较展现了更小的组内变异和更显著的组间差异,有效捕捉到了来自遗传差异较大分支的群落变化。对于ITS这类高度变异、无法构建可信域范围系统树的标记基因,k-mer多样性几乎成为融入序列组成信息的唯一便捷方案。
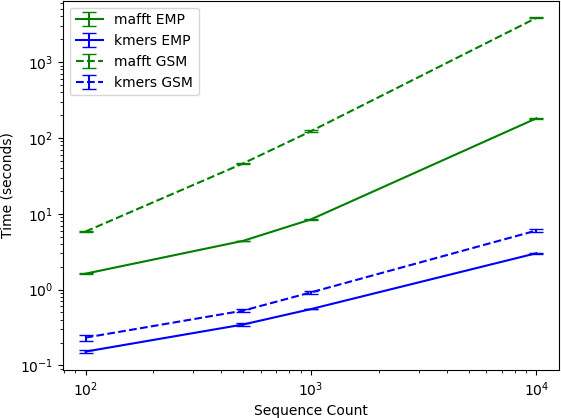
夸张的性价比:毫秒级vs小时级
运行时间实验显示,k-mer计数几乎不随序列长度和数量剧烈增长 。处理10,000条150 bp的V4序列仅需数秒,处理同样数量约800 bp的ITS序列也仅是从秒级增至十秒级。而作为对比的mafft序列比对结合FastTree建树流程,在相同数据量下耗时从数分钟延长至超过一小时。如果将原数据集全部数十万条唯一序列纳入,k-mer计数的速度优势将更加压倒性。
需要注意的误区
论文明确指出k-mer多样性是现有生态学指标的补充而非替代 。k-mer丰富度反映的是群落所含的遗传子序列多样性,而非物种或ASV数目,不适合直接换算成物种数量。当群落拥有少量但系统发育差异巨大的物种时,可能出现物种丰富度低而k-mer丰富度高的反差情形。因此,建议同时报告经典指标和k-mer指标,通过两者的对比获取更立体的群落组成认知------例如,当传统Jaccard距离较大而k-mer Jaccard距离较小时,暗示样本间差异可能主要由近缘菌株的更替(细微的SNP差异)驱动。
5. 软件使用方法
q2-kmerizer作为QIIME 2的插件,安装十分简便。如果你已经拥有QIIME 2 amplicon分发版(版本≥2024.10),只需激活对应conda环境并执行:
bash
pip install q2_kmerizer@git+https://github.com/bokulich-lab/q2-kmerizer.git@main
qiime dev refresh-cache安装完成后,在终端输入qiime kmerizer --help能看到插件描述。
实测环节可以选用论文示例或QIIME 2官方教程数据,例如帕金森病小鼠模型的16S数据。首先下载必要文件:
bash
wget https://data.qiime2.org/2024.10/tutorials/pd-mice/sample_metadata.tsv
wget https://docs.qiime2.org/2024.10/data/tutorials/pd-mice/dada2_table.qza
wget https://docs.qiime2.org/2024.10/data/tutorials/pd-mice/dada2_rep_set.qza基础k-mer计数
只需一条命令即可将ASV丰度表转化为k-mer频率表:
bash
qiime kmerizer seqs-to-kmers \
--i-sequences dada2_rep_set.qza \
--i-table dada2_table.qza \
--o-kmer-table kmer_table.qza \
--p-max-features 5000这里--p-max-features 5000限定了保留最重要的5000个k-mer,适合控制特征维度。
一键化核心多样性分析
插件还提供了一个便捷的core-metrics管道,能够一次性完成计数、抽平、多样性计算并输出交互式散点图:
bash
qiime kmerizer core-metrics \
--i-sequences dada2_rep_set.qza \
--i-table dada2_table.qza \
--p-sampling-depth 1000 \
--m-metadata-file sample_metadata.tsv \
--p-color-by donor \
--p-max-features 5000 \
--output-dir core-metrics/抽平深度(--p-sampling-depth)需根据你的数据实际情况设定。输出目录中将包含基于k-mer频率计算的各类距离矩阵和PCoA可视化文件,可直接用于后续统计检验。
监督学习无缝衔接
生成的k-mer特征表(kmer_table.qza)与QIIME 2生态完全兼容,可以像普通特征表一样喂给q2-sample-classifier:
bash
qiime sample-classifier classify-samples \
--i-table kmer_table.qza \
--m-metadata-file sample_metadata.tsv \
--m-metadata-column donor \
--output-dir sample-classifier/这将利用k-mer特征训练随机森林分类器并评估预测准确度。
实战贴士
进行beta多样性分析时,可以尝试同时开启TF-IDF加权(默认即为CountVectorizer,可在参数中切换),并与ASV、UniFrac的结果平行比较。若分析的是ITS或其它高变区域,建议将k-mer长度设为16或稍高,并通过--p-max-features适当扩大允许的特征数(如10000--20000),以充分捕获序列变异信息。一直要牢记,若计划进行alpha多样性分析,切勿随意使用TF-IDF加权和过于严苛的min_df过滤,否则你的k-mer丰富度将反映不出真实遗传多样性。
6. 文章的引用格式
Bokulich NA. Integrating sequence composition information into microbial diversity analyses with k-mer frequency counting. mSystems. 2025, 10(3): e01550-24. DOI: https://doi.org/10.1128/msystems.01550-24